WO2014123242A1 - 巨核球及び血小板の製造方法 - Google Patents
巨核球及び血小板の製造方法 Download PDFInfo
- Publication number
- WO2014123242A1 WO2014123242A1 PCT/JP2014/053087 JP2014053087W WO2014123242A1 WO 2014123242 A1 WO2014123242 A1 WO 2014123242A1 JP 2014053087 W JP2014053087 W JP 2014053087W WO 2014123242 A1 WO2014123242 A1 WO 2014123242A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- cells
- expression
- cell
- hematopoietic progenitor
- Prior art date
Links
Images














Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0644—Platelets; Megakaryocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/19—Platelets; Megacaryocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/28—Bone marrow; Haematopoietic stem cells; Mesenchymal stem cells of any origin, e.g. adipose-derived stem cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/125—Stem cell factor [SCF], c-kit ligand [KL]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/145—Thrombopoietin [TPO]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/48—Regulators of apoptosis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/73—Hydrolases (EC 3.)
- C12N2501/734—Proteases (EC 3.4.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2502/00—Coculture with; Conditioned medium produced by
- C12N2502/13—Coculture with; Conditioned medium produced by connective tissue cells; generic mesenchyme cells, e.g. so-called "embryonic fibroblasts"
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/11—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from blood or immune system cells
Definitions
- the present invention relates to a method for producing megakaryocytes and platelets from hematopoietic progenitor cells, a method for selecting hematopoietic progenitor cells suitable for producing megakaryocytes, and the like.
- platelets which are essential for blood clotting and hemostasis, are one of the most important blood cells. Platelets are in great demand for leukemia, bone marrow transplantation, anticancer treatment, etc., and the need for a stable supply is high. So far, platelets have been secured not only by a method of collecting blood by donating blood from a donor, but also by a method of administering a TPO-like structural mimetic preparation, a method of differentiating megakaryocytes from umbilical cord blood or bone marrow cells. Recently, a technique for preparing blood cells such as platelets by inducing differentiation of pluripotent stem cells such as ES cells or iPS cells in vitro has also been developed.
- Patent Document 1 Non-Patent Document 1
- Patent Document 2 Patent Document 2
- the inventors prepared the megakaryocyte progenitor cell line immortalized based on stem cells for quantitative problems such as platelets prepared from stem cells, and prepared platelets in vitro.
- an important technique was developed (Patent Document 3).
- Bcl-xl an apoptosis-inhibiting gene, has been successfully matured by forced expression in the megakaryocyte production process (Patent Document 4).
- megakaryocytes form pseudopodial formations called proplatelets (platelet precursors), fragment their cytoplasm and release platelets.
- Megakaryocytes are thought to become multinucleated by endomitosis before releasing platelets.
- Meganuclear cell nuclear fission is multipolar mitosis due to abnormalities of fission and cytokinesis, without fission formation and spindle elongation, resulting in the formation of cells with several lobulated nuclei Is done. Such intranuclear fission occurs repeatedly, which induces multinucleation of megakaryocytes.
- Non-patent Document 1 shows that, in the nuclear division of megakaryocytes, the division groove is formed, but the localization of the non-myocyte myosin II to the contractile ring is not observed, and the contraction ring formation and the spindle elongation. It was clarified that there was a defect. And it was shown that these abnormalities of contraction rings and spindle elongation become more prominent by inhibiting the activity of RhoA and Rock (Non-patent Document 2).
- RhoA accumulates in the division groove and promotes activation of several effector factors including Rho kinase (Rock), citron kinase, LIM kinase and mDia / formins. From these results, it is suggested that the nuclear division of megakaryocytes is promoted by inhibiting the activity of factors involved in the formation of contraction rings such as RhoA and Rock. In addition, there is a report that if the signal of Rho located downstream of integrin alpha2 / beta1 is enhanced, the formation of proplatelet of immature, non-nucleated megakaryocytes is inhibited.
- Non-Patent Document 5 culturing immature megakaryocytes at 39 ° C, higher than the normal culture temperature, promotes the induction of multinucleated mature megakaryocytes and the formation of proplatelets.
- the present inventors have stably used more functional platelets (characterized as CD42b +, which are platelets that retain in vivo activity such as hemostasis) than conventional methods. It was found that it was necessary to establish a megakaryocyte strain that produced more, and in order to overcome this problem, it was thought that the megakaryocyte strain obtained by the conventional method needs to be further matured.
- CD42b + functional platelets
- the present invention aims to provide a method for stopping and maturing the proliferation of megakaryocytes, and a method for selecting a raw material suitable for producing such megakaryocytes.
- the present inventors find a difference between hematopoietic progenitor cells prepared from pluripotent stem cells (ES cells, iPS cells, etc.) and cells that are suitable for establishing megakaryocytes. I tried to do that. Furthermore, in order to mature megakaryocytes, an attempt was made to stop the forced expression of genes required for induction from hematopoietic progenitor cells to megakaryocytes.
- pluripotent stem cells ES cells, iPS cells, etc.
- KLF1 and FLI1 are markers of hematopoietic progenitor cells that can easily establish megakaryocytes by trial and error.
- megakaryocytes Furthermore, in the establishment of megakaryocytes, it was confirmed that the function of megakaryocytes can be maintained by stopping the forced expression of essential genes. Furthermore, the present inventors have found that megakaryocytes whose proliferation has been stopped by stopping gene expression in this way produce functional platelets more efficiently, thereby completing the present invention.
- the present invention [1] A method for producing megakaryocytes from hematopoietic progenitor cells, comprising the following steps (i) to (ii): (i) a step of forcibly expressing an apoptosis inhibitor gene and an oncogene in hematopoietic progenitor cells and culturing; and (ii) A step of culturing the cells obtained in step (i) after stopping the forced expression of apoptosis-suppressing genes and oncogenes.
- step (i) one gene selected from the group consisting of a gene that suppresses expression of p16 gene or p19 gene, a gene that suppresses expression of Ink4a / Arf gene, and a polycomb gene is further added to a hematopoietic progenitor cell Forcibly expressing one gene selected from the group consisting of a gene that suppresses the expression of the p16 gene or the p19 gene, a gene that suppresses the expression of the Ink4a / Arf gene, and a polycomb gene in the step (ii).
- the method according to [1] wherein expression is stopped and culture is performed.
- step (i) a gene selected from the group consisting of an oncogene, a gene that suppresses the expression of the p16 gene or the p19 gene, a gene that suppresses the expression of the Ink4a / Arf gene, and a polycomb gene.
- step (i) a gene selected from the group consisting of an oncogene, a gene that suppresses the expression of the p16 gene or the p19 gene, a gene that suppresses the expression of the Ink4a / Arf gene, and a polycomb gene;
- [5] The method according to any one of [1] to [4], wherein the apoptosis-suppressing gene is a BCL-XL gene.
- [6] The method according to any one of [1] to [5], wherein the oncogene is a c-MYC gene.
- One gene selected from the group consisting of a gene that suppresses the expression of the p16 gene or the p19 gene, a gene that suppresses the expression of the Ink4a / Arf gene, and a polycomb gene is BMI1, [1] to [ [6] The method according to any one of [6].
- [8] The method according to any one of [1] to [7], wherein in the steps (i) and (ii), the cells are cultured on C3H10T1 / 2 cells in a culture solution containing TPO. .
- a method for producing megakaryocyte progenitor cells from hematopoietic progenitor cells comprising the following steps (I) to (II): (I) one gene selected from the group consisting of an oncogene and a gene that suppresses the expression of p16 gene or p19 gene, a gene that suppresses the expression of Ink4a / Arf gene, and a polycomb gene is forcibly expressed in hematopoietic progenitor cells Culturing, and (II) A step of forcibly expressing an apoptosis-suppressing gene in the cells obtained in step (I) or culturing in a medium to which a caspase inhibitor is added.
- apoptosis-suppressing gene is a BCL-XL gene.
- the oncogene is a c-MYC gene.
- the caspase inhibitor is Z-DEVD-FMK.
- One gene selected from the group consisting of the gene that suppresses the expression of the p16 gene or the p19 gene, the gene that suppresses the expression of the Ink4a / Arf gene, and the polycomb gene is BMI1, [20] to [ [23] The method according to any one of [23].
- a gene selected from the group consisting of a gene that suppresses the expression of an exogenous p16 gene or p19 gene that is responsive to a drug, a gene that suppresses the expression of an Ink4a / Arf gene, and a polycomb gene is further chromosome
- One gene selected from the group consisting of the gene that suppresses the expression of the p16 gene or the p19 gene, the gene that suppresses the expression of the Ink4a / Arf gene, and a polycomb gene is BMI1, [27] to [ 30] The cell according to any one of 30.
- a method for selecting hematopoietic progenitor cells suitable for producing megakaryocytes comprising measuring KLF1 expression or FLI1 expression.
- the method according to [32] comprising a step of selecting hematopoietic progenitor cells having low KLF1 expression.
- the method according to [33] comprising a step of selecting hematopoietic progenitor cells having high FLI1 expression.
- hematopoietic progenitor cells are cells induced to differentiate from pluripotent stem cells.
- a method for selecting pluripotent stem cells suitable for megakaryocyte production including the following steps; (I) producing hematopoietic stem cells from pluripotent stem cells; and (Ii) A step of measuring the expression of KLF1 and the expression of FLI1 in the hematopoietic progenitor cells produced in step (i). About.
- megakaryocytes suitable for platelet production can be produced. Moreover, according to the present invention, it is possible to select hematopoietic progenitor cells suitable for producing megakaryocytes.
- an appropriate hematopoietic progenitor cell is selected according to the method of the present invention.
- the resulting megakaryocytes are matured to produce platelets, thereby making it possible to more reliably produce functional platelets from stem cells.
- the obtained platelets are CD42b positive and greatly contribute to clinical application.
- FIG. 1A shows the growth curves of each megakaryocyte strain (TKDN SeV2 Clone-1 to Clone-6) from the 12th day after infection with c-MYC and BCL-XL drug response expressing lentiviruses.
- FIG. 1B shows the results of an expression analysis of KLF1 (left figure) or FLI1 (right figure) in hematopoietic progenitor cells derived from khES3 and TKDN ⁇ SeV2.
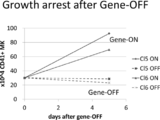
- Figure 2 shows that gene expression was stopped in megakaryocyte strains (Clone-5 (Cl5) and Clone-6 (Cl6)) 18 days after infection with c-MYC and BCL-XL drug-responsive expression lentiviruses.
- FIG. 3 shows the results of flow cytometry of megakaryocytes (Gene-ON) immediately after the forced gene expression cessation and megakaryocytes (Gene-OFF) surface antigens (CD41a and CD42b) 5 days after the cessation.
- the right figure shows the mean fluorescence intensity (MFI) of CD42a (dark ash) and CD42b (light ash), which are surface markers in each megakaryocyte strain. In the figure, it is indicated by an arrow that each marker increases by stopping the forced expression of the gene.
- MFI mean fluorescence intensity
- FIG. 4 shows the results of flow cytometry of surface antigens (CD41a and CD42b) of platelets (Gene-ON) immediately after cessation of forced gene expression and platelets (Gene-OFF) 5 days after cessation.
- the right figure shows the mean fluorescence intensity (MFI) of CD42a (dark ash) and CD42b (light ash), which are surface markers in each platelet. In the figure, it is indicated by an arrow that each marker increases by stopping the forced expression of the gene.
- FIG. 5A shows the internalization of each iPS cell-derived megakaryocyte strain (Clone-1 (Cl1) to Clone-6 (Cl6)) immediately after the forced expression of the gene is stopped (ON) and after 3 days (OFF).
- FIG. 5B shows the endogenous levels of each of the iPS cell-derived megakaryocyte strains (Clone-1 (Cl1) to Clone-6 (Cl6)) immediately after gene expression cessation (ON) and after 3 days after gene expression cessation (OFF).
- the expression levels of exogenous GATA1, p45, b1-tubulin and MPL are shown.
- FIG. 6 shows a megakaryocyte strain established by the method of the present invention (cultured on the 40th day) (upper figure) and a megakaryocyte established by the method described in Takayama et al., Blood, 111: 5298-5306 2008 which is a control cell ( On day 21) (below), the degree of APC-fibrinogen binding (X axis) before (left) and after (right) stimulation with Phorbol 12-Myristate 13-acetate (PMA) is shown.
- PMA Phorbol 12-Myristate 13-acetate
- FIG. 7A shows the result of counting CD41a positive cells obtained from megakaryocyte progenitor cells produced by introducing each gene with respect to the number of culture days.
- FIG. 7B shows a May-Giemsa stained image of megakaryocyte progenitor cells obtained by introducing c-MYC and BMI1.
- FIG. 7C shows a schematic diagram of c-MYC-2A-BMI1 and BMI1-2A- c-MYC expression retroviral vectors.
- FIG. 7D shows the results of counting CD41a positive cells obtained from megakaryocyte progenitor cells produced by introducing each gene with respect to the number of days of culture.
- FIG. 7B shows a May-Giemsa stained image of megakaryocyte progenitor cells obtained by introducing c-MYC and BMI1.
- FIG. 7C shows a schematic diagram of c-MYC-2A-BMI1 and BMI1-2A- c-MYC expression retroviral vectors
- FIG. 7E shows the results of measuring the amount of c-Myc protein when c-MYC and BMI1 were introduced, when c-MYC-2A-BMI1 was introduced, and when BMI1-2A-c-MYC was introduced, respectively.
- FIG. 8A shows the results of counting CD41a positive cells obtained by introducing c-MYC-DD-2A-BMI1 (w DD) or c-MYC -2A-BMI1 (w / o DD) into hematopoietic progenitor cells. Show.
- FIG. 8A shows the results of counting CD41a positive cells obtained by introducing c-MYC-DD-2A-BMI1 (w DD) or c-MYC -2A-BMI1 (w / o DD) into hematopoietic progenitor cells. Show.
- FIG. 8B shows the results of counting CD41a-positive cells on day 7 cultured in a medium to which c-MYC-DD-2A-BMI1 was introduced and each concentration of Shield-1 was added.
- FIG. 8C shows the results of measuring the activity of caspase 3/7 on day 2 cultured in a medium containing c-MYC-DD-2A-BMI1 and each concentration of Shield-1.
- FIG. 9A shows a protocol for producing megakaryocyte progenitor cells by introducing c-MYC, BMI1 and BCL-XL, or c-MYC and BMI1.
- FIG. 9B shows the result of expansion culture of a megakaryocyte progenitor cell line (Cl-1) produced according to the protocol of FIG. 9A.
- FIG. 9C shows the result of expansion culture of a megakaryocyte progenitor cell line (Cl-2) produced according to the protocol of FIG. 9A.
- FIG. 9D shows the results of measuring the amount of c-Myc protein after c-MYC-DD-2A- BMI1 was introduced and cultured in a medium supplemented with each concentration of Shield-1.
- FIG. 9E shows the results of counting CD41a positive cells on day 7 cultured in a medium into which c-MYC-DD-2A-ABMI1 was introduced and each concentration of Shield-1 was added.
- FIG. 10A shows megakaryocytes when c-MYC-2A-BMI1 and BCL-XL are introduced, or when c-MYC-2A-BMI1 is introduced and cultured in a medium supplemented with DMSO or Z-VAD-FMK. The increase rate of progenitor cells is shown.
- FIG. 10B shows the result of expansion culture of a megakaryocyte progenitor cell line (Cl-3) produced by simultaneously introducing c-MYC, BMI1 and BCL-XL.
- FIG. 10C shows the result of expansion culture of a megakaryocyte progenitor cell line (Cl-4) produced by simultaneously introducing c-MYC, BMI1 and BCL-XL.
- FIG. 10D shows the result of expansion culture of a megakaryocyte progenitor cell line (Cl-6) produced by simultaneously introducing c-MYC, BMI1 and BCL-XL.
- FIG. 10E shows the result of expansion culture of a megakaryocyte progenitor cell line (Cl-7) produced by simultaneously introducing c-MYC, BMI1 and BCL-XL.
- FIG. 10F shows the survival of mice administered megakaryocyte progenitor cell line (Cl-1), megakaryocyte progenitor cell line (Cl-7), HL-60 (megakaryocyte cell line) or Meg01 (megakaryocyte cell line). A Kaplan-Meier curve showing the rate is shown.
- FIG. 10E shows the result of expansion culture of a megakaryocyte progenitor cell line (Cl-6) produced by simultaneously introducing c-MYC, BMI1 and BCL-XL.
- FIG. 10F shows the survival of mice administered megakaryocyte progenit
- FIG. 11A shows the number of CD41a positive cells on day 0 or day 21 after freezing and thawing megakaryocyte progenitor cell line (Cl-1) or megakaryocyte progenitor cell line (Cl-2).
- FIG. 11B shows the results of flow cytometry for measuring CD41a, CD42a, CD42b and CD9 after freezing and thawing of a megakaryocyte progenitor cell line (Cl-1).
- FIG. 12A shows a protocol in which expression of a foreign gene is stopped from a megakaryocyte progenitor cell produced by introducing c-MYC, BMI1, and BCL-XL to mature into a megakaryocyte (produce platelets).
- FIG. 12B shows the Giemsa-stained image (upper figure) and the DNA content of the cell (ON) before the cell (OFF) gene expression stop obtained according to the protocol of FIG. 12A.
- FIG. 12C shows a megakaryocyte progenitor cell line (Cl-2 or Cl-7) (OFF) obtained after the protocol of 12A before the gene expression is stopped (Cl-2 or Cl-7) (ON). Shows the results of flow cytometry for CD41a and CD42b.
- FIG. 13A shows CD41a positive / CD42b after expression (OFF) or maintenance (ON) of foreign c-MYC, BMI1 and BCL-XL in megakaryocyte progenitor cell lines (Cl-2 or Cl-7). The increase rate of positive cells is shown.
- FIG. 13B shows the number of CD41a positive / CD42b positive cells when c-MYC, BMI1 and BCL-XL are maintained, only BCL-XL is maintained, and all the expression is stopped.
- FIG. 13C shows the increase rate of CD41a positive / CD42b positive cells when the expression of c-MYC, BMI1 and BCL-XL is maintained and when the existing megakaryocyte strains (CMK, Meg-01 and K562) are stimulated with PMA.
- FIG. 13D shows the expression per ml of culture medium after stopping (OFF) or maintaining (ON) exogenous c-MYC, BMI1 and BCL-XL of megakaryocyte progenitor cell lines (Cl-2 or Cl-7).
- FIG. 14A shows a transmission electron microscope image of platelets produced from megakaryocyte progenitor cell line (Cl-7) or collected blood.
- FIG. 14B shows the results of flow cytometry for CD42a and bound PAC-1 in the case of no stimulation (No stimulation) or thrombin stimulation (Thrombin) of the megakaryocyte progenitor cell line.
- FIG. 14C shows that platelets collected (Fresh platelets), platelets stored at 37 ° C. for 5 days (pooled platelets) and platelets derived from megakaryocyte progenitor cell lines (imMKCL platelets) were not stimulated, or with ADP or thrombin. The binding strength of PAC-1 when stimulated is shown.
- FIG. 14A shows a transmission electron microscope image of platelets produced from megakaryocyte progenitor cell line (Cl-7) or collected blood.
- FIG. 14B shows the results of flow cytometry for CD42a and bound PAC-1 in the case of no stimulation (No stimulation) or thro
- FIG. 14D shows the results of measurement of aggregated platelets (CD9-APC positive / CD9-Pacific Blue positive) when stimulation was not performed with Fresh platelets or imMKCL platelets, or when stimulation was performed with ADP and TRAP.
- FIG. 14D shows the aggregated platelet content (left panel) and even aggregated platelet content (right panel) upon Collaegen stimulation as measured in FIG. 14D.
- FIG. 14F shows a microscope image obtained by observing platelet aggregates derived from Cl-7 or Cl-2 under a flow rate of 1600S ⁇ 1 (direction is shown at the top).
- FIG. 14G shows the number of agglomerates observed in FIG. 14F.
- FIG. 15A and B show 30 minutes, 2 hours or 30 minutes after administration of platelets from megakaryocyte progenitor cell lines (6 ⁇ 10 8 or 1 ⁇ 10 8 ) or blood collected platelets (1 ⁇ 10 8 ) to mice.
- the content rate in blood in 24 hours (Human CD41a positive / Mouse CD41 negative) is shown.
- FIG. 15C shows a confocal microscope image obtained by imaging the time-dependent movement of blood vessels (red) of platelets (green) derived from megakaryocyte precursor cell lines in vivo.
- FIG. 15D shows the number of platelets derived from megakaryocyte progenitor cell lines adhered per 100 ⁇ m blood vessel.
- FIG. 15E shows a microscopic image of platelets (green) derived from megakaryocyte progenitor cell lines in thrombus (20 sec) generated at the site of laser irradiation damage.
- FIG. 15E shows the following after administration of blood collected platelets (Fresh), stored platelets (pooled), and platelets derived from megakaryocyte progenitor cell lines (Cl-1, Cl-2, Cl-3 and Cl-7) The number of platelets contained in the thrombus is shown.
- the present invention provides a method for producing megakaryocytes from hematopoietic progenitor cells.
- One aspect of the method for producing megakaryocytes according to the present invention includes a step of forcibly expressing apoptosis-suppressing genes and oncogenes in hematopoietic progenitor cells and culturing the cells, and suspending the forced expression of apoptosis-inhibiting genes and oncogenes The process of carrying out is included.
- a cell obtained by forcibly expressing an apoptosis-inhibiting gene and an oncogene and culturing the cell may be a megakaryocyte progenitor cell.
- the “megakaryocyte” in the present invention may be a multinucleated cell, and includes, for example, a cell characterized as CD41a positive / CD42a positive / CD42b positive.
- the cells may be characterized as cells expressing GATA1, FOG1, NF-E2, and ⁇ 1-tubulin.
- a multinucleated megakaryocyte refers to a cell or a group of cells in which the number of nuclei is relatively increased as compared to hematopoietic progenitor cells. For example, when the nuclei of hematopoietic progenitor cells to which the method of the present invention is applied is 2N, 4N or more cells become multinucleated megakaryocytes.
- the megakaryocyte may be immortalized as a megakaryocyte strain or may be a group of cloned cells.
- the “megakaryocyte progenitor cell” in the present invention is a cell that becomes a megakaryocyte upon maturation and is not multinucleated, for example, a cell characterized as CD41a positive / CD42a positive / CD42b weak positive. Including.
- the megakaryocyte progenitor cells of the present invention are preferably cells that can be expanded by expansion culture, for example, cells that can be expanded under appropriate conditions for at least 60 days or longer.
- the megakaryocyte progenitor cell may or may not be cloned, and is not particularly limited, but the cloned cell may be referred to as a megakaryocyte progenitor cell line.
- hematopoietic progenitor cells are cells that can differentiate into blood cells such as lymphocytes, eosinophils, neutrophils, basophils, erythrocytes, megakaryocytes, etc. Hematopoietic stem cells are not distinguished and indicate the same cells unless otherwise specified. Hematopoietic stem / progenitor cells can be recognized by, for example, positive surface antigens CD34 and / or CD43. In the present invention, hematopoietic stem cells can also be applied to pluripotent stem cells, hematopoietic progenitor cells derived from cord blood, bone marrow blood, peripheral blood-derived hematopoietic stem cells and progenitor cells.
- pluripotent stem cells are cultured on C3H10T1 / 2 in the presence of VEGF according to the method described in Takayama N., et al. J Exp Med. 2817-2830 (2010) Can be prepared from a net-like structure (also referred to as ES-sac or iPS-sac).
- the “net-like structure” is a three-dimensional sac-like structure (with space inside) derived from pluripotent stem cells, which is formed by an endothelial cell population and the like, and contains hematopoietic progenitor cells inside. It is a structure.
- hematopoietic progenitor cells from pluripotent stem cells
- embryoid bodies are formed and cytokines are added (Chadwick et al. Blood 2003, 102: ⁇ 906-15, Vijayaravavan et al. Cell Stem Cell 2009, 4: 248-62, Saeki et al. Stem Cells 2009, 27: 59-67) or co-culture with heterologous stromal cells (Niwa A et al. J Cell Physiol. 2009 Nov; 221 (2 ): 367-77.) And the like.
- a preferred hematopoietic progenitor cell in the present invention is a hematopoietic progenitor cell with low expression of KLF1 gene or high expression of FLI1 gene. Therefore, when producing megakaryocytes, a step of selecting hematopoietic progenitor cells with low KLF1 expression or high FLI1 expression may be included.
- the low expression of KLF1 means that the expression of KLF1 is low compared to the control, and the control is not particularly limited, and can be appropriately selected by those skilled in the art based on literature or experience.
- hematopoietic progenitor cells produced from khES3 according to the method described in Takayama N., et al. J Exp Med. 2817-2830 (2010) are exemplified.
- KLF1 is a gene described in NCBI accession number NM_006563.
- a low expression of FLI1 means that the expression of FLI1 is low compared to the control, and the control is not particularly limited and may be appropriately selected by those skilled in the art based on literature or experience.
- the control is not particularly limited and may be appropriately selected by those skilled in the art based on literature or experience.
- hematopoietic progenitor cells produced from khES3 according to the method described in Takayama N., et al. J Exp Med. 2817-2830 (2010) are exemplified.
- FLI1 is a gene described in NCBI accession numbers NM_001167681, NM_001271010, NM_001271012, or NM_002017.
- a method for measuring gene expression it can be performed according to a conventional method of those skilled in the art, and examples thereof include a DNA chip method, a Southern blot method, a Northern blot method, an RT-PCR (Polymerase Chain Reaction) method, and the like.
- in vivo hematopoietic progenitor cells may be used as a control.
- any two or more types of hematopoietic progenitor cells (group) may be compared to select those having lower KLF1 expression or higher FLI1 expression.
- the present invention selects the hematopoietic progenitor cells produced at the same time with lower KLF1 expression or higher FLI1 expression. You may use for.
- the expression of low or high expression is not limited to the case where the expression is significantly low or high compared to the control, and includes cases where the expression is low or high enough to be recognized by those skilled in the art as having a tendency to be low or high.
- a pluripotent stem cell is a stem cell that has pluripotency that can be differentiated into all cells present in a living body and also has a proliferative ability, and includes, for example, an embryonic stem (ES) cells, embryonic stem (ntES) cells derived from cloned embryos obtained by nuclear transfer, sperm stem cells (“GS cells”), embryonic germ cells (“EG cells”), induced pluripotent stem (iPS) cells.
- GS cells embryonic stem cells
- EG cells embryonic germ cells
- iPS induced pluripotent stem
- cultured fibroblasts cultured fibroblasts, bone marrow stem cell-derived pluripotent cells (Muse cells), stimulation-induced pluripotent acquisition cells (STAP cells), and the like are included.
- ES cells are stem cells established from the inner cell mass of early embryos (for example, blastocysts) of mammals such as humans and mice, and having pluripotency and proliferation ability by self-replication.
- ES cells are embryonic stem cells derived from the inner cell mass of the blastocyst, the embryo after the morula, in the 8-cell stage of a fertilized egg, and have the ability to differentiate into any cell that constitutes an adult, so-called differentiation. And ability to proliferate by self-replication.
- ES cells were discovered in mice in 1981 (MJ Evans and MH Kaufman (1981), Nature 292: 154-156), and then ES cell lines were also established in primates such as humans and monkeys (JA Thomson et al.
- ES cells can be established by taking an inner cell mass from a blastocyst of a fertilized egg of a target animal and culturing the inner cell mass on a fibroblast feeder. In addition, maintenance of cells by subculture is performed using a culture solution to which substances such as leukemia inhibitory factor (LIF) and basic fibroblast growth factor (basic fibroblast growth factor (bFGF)) are added. It can be carried out.
- LIF leukemia inhibitory factor
- bFGF basic fibroblast growth factor
- DMEM / F-12 culture medium supplemented with 0.1 mM 2-mercaptoethanol, 0.1 mM non-essential amino acid, 2 mM L-glutamic acid, 20% KSR and 4 ng / ml bFGF is used as the culture medium for ES cell production.
- Human ES cells can be maintained in a humid atmosphere at 37 ° C., 5% CO 2 (H. Suemori et al. (2006), Biochem. Biophys. Res. Commun., 345: 926-932).
- ES cells also need to be passaged every 3-4 days, where passage is eg 0.25% trypsin and 0.1 mg / ml collagenase IV in PBS containing 1 mM CaCl 2 and 20% KSR. Can be used.
- ES cells can be generally selected by Real-Time PCR using the expression of gene markers such as alkaline phosphatase, Oct-3 / 4, Nanog as an index.
- gene markers such as alkaline phosphatase, Oct-3 / 4, Nanog
- OCT-3 / 4, NANOG, and ECAD can be used as an index (E. Kroon et al. (2008), Nat. Biotechnol., 26: 443). -452).
- Human ES cell lines for example, WA01 (H1) and WA09 (H9) are obtained from the WiCell Research Institute, and KhES-1, KhES-2 and KhES-3 are obtained from the Institute of Regenerative Medicine, Kyoto University (Kyoto, Japan) Is possible.
- sperm stem cells are testis-derived pluripotent stem cells that are the origin of spermatogenesis. Like ES cells, these cells can be induced to differentiate into various types of cells, and have characteristics such as the ability to create chimeric mice when transplanted into mouse blastocysts (M. Kanatsu-Shinohara et al. ( 2003) Biol. Reprod., 69: 612-616; K. Shinohara et al. (2004), Cell, 119: 1001-1012).
- GDNF glial cell line-derived neurotrophic factor
- Embryonic germ cells are cells that are established from embryonic primordial germ cells and have the same pluripotency as ES cells, such as LIF, bFGF, stem cell factor, etc. It can be established by culturing primordial germ cells in the presence of these substances (Y. Matsui et al. (1992), Cell, 70: 841-847; JL Resnick et al. (1992), Nature, 359: 550 -551).
- iPS Artificial pluripotent stem cells
- somatic cells in the form of DNA or protein, which is almost equivalent to ES cells
- It is an artificial stem cell derived from a somatic cell having the characteristics of, for example, differentiation pluripotency and proliferation ability by self-replication (K. Takahashi and S. Yamanaka (2006) Cell, 126: 663-676; K. Takahashi et al (2007), Cell, 131: 861-872; J. Yu et al. (2007), Science, 318: 1917-1920; Nakagawa, M. et al., Nat. Biotechnol.
- the reprogramming factor is a gene specifically expressed in ES cells, its gene product or non-cording RNA, a gene that plays an important role in maintaining undifferentiation of ES cells, its gene product or non-coding RNA, or It may be constituted by a low molecular compound.
- genes included in the reprogramming factor include Oct3 / 4, Sox2, Sox1, Sox3, Sox15, Sox17, Klf4, Klf2, c-Myc, N-Myc, L-Myc, Nanog, Lin28, Fbx15, ERas, ECAT15 -2, Tcl1, beta-catenin, Lin28b, Sall1, Sall4, Esrrb, Nr5a2, Tbx3 or Glis1 etc. are exemplified, and these reprogramming factors may be used alone or in combination.
- the reprogramming factors include histone deacetylase (HDAC) inhibitors [for example, small molecule inhibitors such as valproate (VPA), trichostatin A, sodium butyrate, MC 1293, M344, siRNA and shRNA against HDAC (eg Nucleic acid expression inhibitors such as HDAC1DACsiRNA Smartpool ⁇ (Millipore), HuSH 29mer shRNA Constructs against HDAC1 (OriGene) etc.], MEK inhibitors (eg PD184352, PD98059, U0126, SL327 and PD0325901), Glycogen synthasekinskin -3 inhibitors (eg, Bio and CHIR99021), DNA methyltransferase inhibitors (eg, 5-azacytidine), histone methyltransferase inhibitors (eg, small molecule inhibitors such as BIX-01294, Suv39hl, Suv39h2, SetDBl and G9a Nucleic acid expression inhibitors such as si
- the reprogramming factor may be introduced into a somatic cell by a technique such as lipofection, fusion with a cell membrane-permeable peptide (for example, HIV-derived TAT and polyarginine), or microinjection.
- a cell membrane-permeable peptide for example, HIV-derived TAT and polyarginine
- Virus vectors include retrovirus vectors, lentivirus vectors (cell, 126, pp.663-676, 2006; Cell, 131, pp.861-872, 2007; Science, 318, pp.1917-1920, 2007 ), Adenovirus vectors (Science, 322, 945-949, 2008), adeno-associated virus vectors, Sendai virus vectors (WO 2010/008054), and the like.
- artificial chromosome vectors examples include human artificial chromosomes (HAC), yeast artificial chromosomes (YAC), and bacterial artificial chromosomes (BAC, PAC).
- HAC human artificial chromosomes
- YAC yeast artificial chromosomes
- BAC bacterial artificial chromosomes
- a plasmid a plasmid for mammalian cells can be used (Science, 322: 949-953, 2008).
- the vector can contain regulatory sequences such as a promoter, enhancer, ribosome binding sequence, terminator, polyadenylation site, etc. so that a nuclear reprogramming substance can be expressed.
- Selective marker sequences such as kanamycin resistance gene, ampicillin resistance gene, puromycin resistance gene, thymidine kinase gene, diphtheria toxin gene, reporter gene sequences such as green fluorescent protein (GFP), ⁇ -glucuronidase (GUS), FLAG, etc.
- GFP green fluorescent protein
- GUS ⁇ -glucuronidase
- FLAG FLAG
- the above vector has a LoxP sequence before and after the introduction of the gene into a somatic cell in order to excise the gene or promoter encoding the reprogramming factor and the gene encoding the reprogramming factor that binds to it. May be.
- RNA it may be introduced into somatic cells by techniques such as lipofection and microinjection, and in order to suppress degradation, RNA incorporating 5-methylcytidine and pseudouridine® (TriLink® Biotechnologies) is used. Yes (Warren L, (2010) Cell Stem Cell. 7: 618-630).
- Examples of the culture medium for inducing iPS cells include DMEM, DMEM / F12 or DME culture medium containing 10 to 15% FBS (these culture media include LIF, penicillin / streptomycin, puromycin, L-glutamine). , Non-essential amino acids, ⁇ -mercaptoethanol, etc.) or commercially available culture media (eg, culture media for mouse ES cell culture (TX-WES culture solution, Thrombo X), primate ES cells) Culture medium for culture (primate ES / iPS cell culture medium, Reprocell), serum-free medium (mTeSR, Stemcell Technology).
- DMEM DMEM / F12 or DME culture medium containing 10 to 15% FBS
- these culture media include LIF, penicillin / streptomycin, puromycin, L-glutamine). , Non-essential amino acids, ⁇ -mercaptoethanol, etc.
- commercially available culture media eg, culture media for mouse ES cell culture (TX
- a somatic cell is brought into contact with a reprogramming factor on a DMEM or DMEM / F12 medium containing 10% FBS at 37 ° C. in the presence of 5% CO 2 for about 4 to 7 days. Then, re-spread the cells on feeder cells (for example, mitomycin C-treated STO cells, SNL cells, etc.), and use bFGF-containing primate ES cell culture medium about 10 days after contact between the somatic cells and the reprogramming factor. Culturing and generating iPS-like colonies about 30 to about 45 days or more after the contact.
- feeder cells for example, mitomycin C-treated STO cells, SNL cells, etc.
- 10% FBS-containing DMEM medium including LIF, penicillin / streptomycin, etc.
- feeder cells eg, mitomycin C-treated STO cells, SNL cells, etc.
- 5% CO 2 at 37 ° C. can be suitably included with puromycin, L-glutamine, non-essential amino acids, ⁇ -mercaptoethanol, etc.
- ES-like colonies after about 25 to about 30 days or more .
- somatic cells to be reprogrammed themselves are used (Takahashi K, et al. (2009), PLoS One. 4: e8067 or WO2010 / 137746), or extracellular matrix (eg, Laminin- 5 (WO2009 / 123349) and Matrigel (BD)) are exemplified.
- iPS cells may be established under hypoxic conditions (oxygen concentration of 0.1% or more and 15% or less) (Yoshida Y, et al. (2009), Cell Stem Cell. 5: 237 -241 or WO2010 / 013845).
- the culture medium is exchanged with a fresh culture medium once a day from the second day onward.
- the number of somatic cells used for nuclear reprogramming is not limited, but ranges from about 5 ⁇ 10 3 to about 5 ⁇ 10 6 cells per 100 cm 2 of culture dish.
- IPS cells can be selected according to the shape of the formed colonies.
- a drug resistance gene that is expressed in conjunction with a gene that is expressed when somatic cells are initialized for example, Oct3 / 4, Nanog
- a culture solution containing the corresponding drug selection The established iPS cells can be selected by culturing with the culture medium.
- the marker gene is a fluorescent protein gene
- iPS cells are selected by observing with a fluorescence microscope, in the case of a luminescent enzyme gene, by adding a luminescent substrate, and in the case of a chromogenic enzyme gene, by adding a chromogenic substrate can do.
- the term “somatic cell” refers to any animal cell (preferably, a mammalian cell including a human) except a germ line cell such as an egg, oocyte, ES cell, or totipotent cell.
- Somatic cells include, but are not limited to, fetal (pup) somatic cells, neonatal (pup) somatic cells, and mature healthy or diseased somatic cells. , Passage cells, and established cell lines.
- somatic cells include, for example, (1) neural stem cells, hematopoietic stem cells, mesenchymal stem cells, tissue stem cells such as dental pulp stem cells (somatic stem cells), (2) tissue progenitor cells, (3) lymphocytes, epithelium Cells, endothelial cells, muscle cells, fibroblasts (skin cells, etc.), hair cells, hepatocytes, gastric mucosal cells, enterocytes, spleen cells, pancreatic cells (exocrine pancreas cells, etc.), brain cells, lung cells, kidney cells Examples thereof include differentiated cells such as fat cells.
- the HLA genotype of the transplant recipient is the same or substantially the same from the viewpoint that rejection is less likely to occur. It is desirable to use somatic cells for the production of iPS cells.
- “substantially the same” means that the HLA genotype matches the transplanted cells to such an extent that an immune response can be suppressed by an immunosuppressive agent.
- HLA-A, HLA-B It may also be a somatic cell having an HLA type in which 3 loci of HLA-DR or 4 loci plus HLA-C are matched.
- E Cloned embryo-derived ES cells obtained by nuclear transfer nt ES cells are cloned embryo-derived ES cells produced by nuclear transfer technology and have almost the same characteristics as ES cells derived from fertilized eggs (T. Wakayama et al. (2001), Science, 292: 740-743; S. Wakayama et al. (2005), Biol. Reprod., 72: 932-936; J. Byrne et al. (2007) , Nature, 450: 497-502).
- an ES cell established from an inner cell mass of a clonal embryo-derived blastocyst obtained by replacing the nucleus of an unfertilized egg with the nucleus of a somatic cell is an nt ES (nuclear transfer ES) cell.
- nt ES nuclear transfer ES
- nuclear transfer technology JB Cibelli et al. (1998), Nature Biotechnol., 16: 642-646)
- ES cell production technology is used (Kiyaka Wakayama et al. ( 2008), Experimental Medicine, Vol.26, No.5 (extra number), 47-52).
- Nuclear transfer can be initialized by injecting a somatic cell nucleus into a mammal's enucleated unfertilized egg and culturing for several hours.
- Muse cells are pluripotent stem cells produced by the method described in WO2011 / 007900. Specifically, fibroblasts or bone marrow stromal cells are treated with trypsin for a long time, preferably 8 or 16 hours. It is a pluripotent cell obtained by suspension culture after treatment, and is positive for SSEA-3 and CD105.
- STAP cells Stimulus-induced pluripotent acquisition cells
- STAP cells are pluripotent stem cells produced by the method described in WO2013 / 163296.
- SSEA-4 is obtained by culturing somatic cells in an acidic solution at pH 5.4 to 5.8 for 30 minutes.
- E-cadherin positive cells are obtained by culturing somatic cells in an acidic solution at pH 5.4 to 5.8 for 30 minutes.
- preferred pluripotent stem cells are cells capable of producing hematopoietic progenitor cells capable of inducing differentiation into megakaryocytes. Selection of such pluripotent stem cells can be performed depending on whether hematopoietic progenitor cells with lower KLF1 expression or higher FLI1 expression can be produced.
- the method for producing hematopoietic progenitor cells from pluripotent stem cells and the method for measuring the expression of KLF1 and the expression of FLI1 can be performed using the methods described above.
- the expression of KLF1 and FLI1 is expressed in (1) the step of producing hematopoietic stem cells from pluripotent stem cells, and (2) the hematopoietic progenitor cells produced in step (1).
- the term “oncogene” refers to a gene that causes canceration of normal cells due to its expression, structure or function being different from that of normal cells.
- MYC family genes MYC family genes, Src family genes , Ras family genes, Raf family genes, protein kinase family genes such as c-Kit, PDGFR, and Abl.
- MYC family genes include c-MYC, N-MYC, and L-MYC. More preferably, it is a c-MYC gene.
- the c-MYC gene is, for example, a gene consisting of a nucleic acid sequence represented by NCBI accession number NM_002467.
- the c-MYC gene may also include homologues thereof.
- the c-MYC gene homologue is a sequence whose cDNA sequence is substantially the same as the nucleic acid sequence represented by NCBI accession number NM_002467, for example. It is a gene consisting of A cDNA consisting of a sequence substantially identical to the nucleic acid sequence shown by NCBI accession number NM_002467 is about 60% or more, preferably about 70%, of the DNA consisting of the sequence shown by NCBI accession number NM_002467.
- stringent conditions are hybridization conditions that are easily determined by those skilled in the art, and are generally empirical experimental conditions that depend on the probe length, washing temperature, and salt concentration. In general, the longer the probe, the higher the temperature for proper annealing, and the shorter the probe, the lower the temperature. Hybridization generally relies on the ability to reanneal in an environment where the complementary strand is slightly below its melting point.
- low stringency conditions include washing in a 0.1 ⁇ SSC, 0.1% SDS solution at a temperature of 37 ° C. to 42 ° C. in the filter washing step after hybridization.
- highly stringent conditions include washing in 65 ° C., 5 ⁇ SSC and 0.1% SDS in the washing step.
- c-MYC encoding a protein fused with a destabilizing domain may be used.
- the destabilizing domain can be purchased from ProteoTuner or Clontech.
- the “apoptosis-suppressing gene” is not particularly limited as long as it is a gene that suppresses apoptosis, and examples thereof include BCL2 gene, BCL-XL gene, Survivin, and MCL1.
- BCL2 gene BCL-XL gene
- BCL-XL gene is a gene consisting of a nucleic acid sequence represented by NCBI accession number NM_001191 or NM_138578, for example.
- the BCL-XL gene may also include a homologue thereof.
- the homologue of the BCL-XL gene is substantially the same as the nucleic acid sequence represented by the NCBI accession number NM_001191 or NM_138578, for example.
- the cDNA comprising substantially the same sequence as the nucleic acid sequence represented by NCBI accession number NM_001191 or NM_138578 is approximately 60% or more, preferably the DNA comprising the sequence represented by NCBI accession number NM_001191 or NM_138578.
- one obtained by forcibly expressing any of the following genes (i) to (iii) in hematopoietic progenitor cells and culturing and proliferating the cells can be used.
- a gene that suppresses expression of the p16 gene or the p19 gene a gene that suppresses the expression of the Ink4a / Arf gene; and
- a gene that suppresses the expression of the Ink4a / Arf gene a gene that suppresses the expression of the Ink4a / Arf gene.
- Polycomb gene Polycomb gene.
- Examples of the genes of (i) to (iii) include BMI1, Mel18, Ring1a / b, Phc1 / 2/3, Cbx2 / 4/6/7/8, Ezh2, Eed, Suz12, HADC, Dnmt1 / 3a / 3b can be mentioned, but the BMI1 gene is particularly preferred.
- the BMI1 gene is, for example, a gene consisting of a nucleic acid sequence represented by NCBI accession number NM_005180.
- the BMI1 gene may also include a homologue thereof.
- the homologue of the BMI1 gene is a gene whose cDNA sequence is substantially the same as the nucleic acid sequence represented by NCBI accession number NM_005180, for example. That is.
- the cDNA consisting of a sequence substantially identical to the nucleic acid sequence shown by NCBI accession number NM_005180 is about 60% or more, preferably about 70%, of the DNA consisting of the sequence shown by NCBI accession number NM_005180.
- DNA complementary to the nucleic acid sequence represented by NCBI accession number NM_005180 DNA that is capable of hybridizing under stringent conditions, and the protein encoded by the DNA suppresses oncogene-induced cell aging that occurs in cells in which oncogenes such as MYC family genes are expressed, It promotes the amplification of the cells.
- a gene that suppresses the expression of the p16 gene or the p19 gene (i) a gene that suppresses the expression of the Ink4a / Arf gene, and (iii) one gene selected from the group consisting of a polycomb gene,
- a method for producing megakaryocytes that further includes a step of stopping and culturing the forcedly expressed gene is preferable.
- this step preferably selected from the group consisting of an oncogene, and (i) a gene that suppresses expression of p16 gene or p19 gene, (ii) a gene that suppresses expression of Ink4a / Arf gene, and (iii) a polycomb gene
- This is a step of forcibly expressing one gene to be expressed in hematopoietic progenitor cells and then forcibly expressing an apoptosis-suppressing gene in the cells.
- the above gene can be forcibly expressed in hematopoietic progenitor cells according to the ordinary method of those skilled in the art.
- vectors expressing these genes, or proteins or RNAs encoding these genes In the form of hematopoietic progenitor cells.
- it can be performed by bringing a low molecular weight compound or the like that induces expression of these genes into contact with hematopoietic progenitor cells.
- an expression vector, protein, RNA or a low molecular weight compound that induces expression should be introduced multiple times in accordance with the necessary period. Can be done.
- vectors expressing these genes include retrovirus, lentivirus, adenovirus, adeno-associated virus, herpes virus, Sendai virus and other viral vectors, animal cell expression plasmids (eg, pA1-11, pXT1, pRc / CMV, pRc / RSV, pcDNAI / Neo) and the like can be used.
- a retroviral vector or a lentiviral vector is preferable in that it can be carried out by single introduction.
- promoters used in expression vectors include EF- ⁇ promoter, CAG promoter, SR ⁇ promoter, SV40 promoter, LTR promoter, CMV (cytomegalovirus) promoter, RSV (rous sarcoma virus) promoter, MoMuLV (Moloney murine leukemia) Virus) LTR, HSV-TK (herpes simplex virus thymidine kinase) promoter, etc. are used.
- the expression vector may optionally contain an enhancer, a poly A addition signal, a selection marker gene, an SV40 replication origin, and the like.
- Useful selection marker genes include, for example, dihydrofolate reductase gene, neomycin resistance gene, puromycin resistance gene and the like.
- the gene expression since the gene expression is controlled by tetracycline or doxycycline, it may be a drug-responsive vector having a tetracycline-responsive element in the promoter region.
- an expression vector in which a loxP sequence is placed so as to sandwich the gene and / or promoter region with the loxP sequence may be used.
- the genes may be vertically linked to obtain a polycistronic vector.
- it is ligated between 2A self-cleaving peptides of foot-and-mouth disease virus (see Science, 322, 949-953, 2008, etc.) and genes that forcefully express IRES sequences, etc. obtain.
- a plasmid containing the nucleic acid is used as an appropriate packaging cell (eg, Plat-E cell) or a complementary cell line (eg, 293 cell). And the virus produced in the culture supernatant is collected and contacted with hematopoietic progenitor cells for infection.
- an appropriate packaging cell eg, Plat-E cell
- a complementary cell line eg, 293 cell
- a plasmid vector in the case of a non-viral vector, can be introduced into a cell using a lipofection method, a liposome method, an electroporation method, a calcium phosphate coprecipitation method, a DEAE dextran method, a microinjection method, a gene gun method, or the like.
- a caspase inhibitor may be brought into contact with cells instead of forcing the apoptosis-suppressing gene to be expressed in hematopoietic progenitor cells.
- the caspase inhibitor may be a peptidic compound, a non-peptidic compound, or a biological protein. Examples of the peptide compound include the following peptide compounds (1) to (10) which are artificially chemically synthesized.
- anilinoquinazolines (AQZs) -AstraZeneca-Pharmaceuticals (Scott et al., J. Pharmacol. Exp. Ther. 304, 433-440 (2003)), (2) M826-Merck Frosst (Han et al., J. Biol. Chem. 277, 30128-30136 (2002)), (3) M867-Merck Frosst (Methot et al., J.Exp. Med. 199, 199-207 (2004)), (4) Nicotinyl aspartyl ketones- Merck Frosst (Isabel et al., Bioorg. Med. Chem. Lett. 13, 2137-2140 (2003)), etc. Can be illustrated.
- IDN-6556-Idun Pharmaceuticals (Hoglen et al., J.Pharmacol. Exp. Ther. 309, 634-640 (2004)), (2) MF-286 and MF-867-Merck Frosst (Los et al., Drug Discov. Today 8, 67-77 (2003)), (3) IDN-5370-Idun Pharmaceuticals (Deckwerth et al., Drug Dev. Res. 52, 579-586 (2001)), (4) IDN-1965-Idun Pharmaceuticals (Hoglen et al., J. Pharmacol. Exp. Ther.
- VX-799- Vertex Pharmaceuticals Lis et al., Drug Drugs, Today 8 and 67-77 (2003).
- M-920 and M-791-Merck Frosst can also be mentioned as caspase inhibitors.
- a preferred caspase inhibitor is Z-VAD FMK, and when Z-VAD FMK is used, it is performed by adding hematopoietic progenitor cells to the culture medium, and the concentration of Z-VAD FMK in the preferred medium Examples include 10 ⁇ M or more, 20 ⁇ M or more, 30 ⁇ M or more, 40 ⁇ M or more, and 50 ⁇ M or more, and preferably 30 ⁇ M or more.
- the present invention as described above, as a method for culturing cells in which an exogenous gene such as an apoptosis-suppressing gene is forcibly expressed, a method of culturing on feeder cells using an arbitrary medium is exemplified.
- the feeder cells are not particularly limited as long as they can induce megakaryocytes or megakaryocyte progenitor cells.
- C3H10T1 / 2 Keratagiri T, et al., Biochem Biophys Res Commun. 172, 295-299 (1990)).
- the medium used in the present invention is not particularly limited, but a medium used for culturing animal cells can be prepared as a basal medium.
- basal media include IMDM medium, MediumMedi199 medium, Eagle's Minimum Essential Medium (EMEM) medium, ⁇ MEM medium, Dulbecco's modified Eagle's Medium (DMEM) medium, Ham's F12 medium, RPMI 1640 medium, Fischer Life's medium, Neurosal's medium And a mixed medium thereof.
- the medium may contain serum or may be serum-free.
- the medium can be, for example, albumin, insulin, transferrin, selenium, fatty acids, trace elements, 2-mercaptoethanol, thiolglycerol, lipids, amino acids, L-glutamine, non-essential amino acids, vitamins, growth factors, small molecules
- One or more substances such as compounds, antibiotics, antioxidants, pyruvate, buffers, inorganic salts, cytokines and the like may also be included.
- Cytokines are proteins that promote blood cell differentiation, and examples include VEGF, TPO, SCF, and the like.
- a preferable medium in the present invention is an IMDM medium containing serum, insulin, transferrin, serine, thiolglycerol, ascorbic acid, and TPO. More preferably, it further contains SCF.
- a drug-responsive promoter it is desirable to include a corresponding drug such as tetracycline or doxycycline in the medium in the forced expression step.
- conditions for culturing are not particularly limited, but it has been confirmed that culturing at a temperature of 37 ° C. or higher promotes differentiation of megakaryocytes or megakaryocyte progenitor cells.
- the temperature of 37 ° C. or higher is appropriate as a temperature that does not damage cells, and is preferably about 37 ° C. to about 42 ° C., preferably about 37 to about 39 ° C., for example.
- the culture period at a temperature of 37 ° C. or higher can be appropriately determined while monitoring the number of megakaryocytes or megakaryocyte progenitor cells.
- the number of days is not particularly limited as long as the desired megakaryocyte progenitor cell is obtained.For example, at least 6 days, 12 days, 18 days, 24 days, 30 days, 42 days, 48 days, 54 days, 54 More than 60 days and more preferably 60 days or more.
- the long culture period is not a problem in the production of megakaryocytes.
- one embodiment of the method for producing megakaryocytes further includes (a) a substance that inhibits the expression or function of the p53 gene product, (b) an actomyosin complex function inhibitor, (c) a ROCK inhibitor, and ( d)
- the medium may further contain an HDAC inhibitor.
- the method for producing megakaryocytes of the present invention further includes a step of suspending forced expression and culturing the megakaryocytes or megakaryocyte precursor cells obtained in the step of forcibly expressing foreign genes as described above.
- a method of stopping forced expression for example, when forced expression is performed using a drug-responsive vector, it may be achieved by not contacting the corresponding drug with the cell.
- the above-mentioned vector containing LoxP it may be achieved by introducing Cre recombinase into the cell.
- a transient expression vector and RNA or protein introduction are used, the contact with the vector or the like may be stopped.
- the medium used in this step can be performed using the same medium as described above.
- the conditions for culturing with the forced expression stopped are not particularly limited, but for example, about 37 ° C. to about 42 ° C., preferably about 37 to about 39 ° C. are preferable.
- the culture period at a temperature of 37 ° C. or higher can be determined as appropriate while monitoring the number of megakaryocytes. For example, it is 2 days to 10 days, preferably about 3 days to 7 days. is there. Desirably at least 3 days. In addition, it is desirable to perform subculture as appropriate during the culture period.
- the megakaryocytes obtained by the above method are sufficiently mature and efficiently produce CD42b-positive functional platelets.
- This CD42b positive platelet has a high thrombus formation ability in vivo and invitro.
- the megakaryocyte obtained in the present invention is a megakaryocyte in which at least an exogenous apoptosis-inhibiting gene and an oncogene are incorporated in the chromosome, but the expression of the gene is stopped.
- maturation of megakaryocytes means that megakaryocytes are sufficiently polynucleated and can produce functional platelets. Megakaryocyte maturation can also be confirmed by, for example, increased expression of megakaryocyte maturation-related genes such as GATA1, p45 NF-E2, and beta1-tubulin.
- megakaryocytes and / or megakaryocyte progenitor cells can produce functional platelets even after thawing after cryopreservation, and therefore megakaryocytes and / or megakaryocytes produced using the method of the present invention Progenitor cells can be distributed in a cryopreserved state.
- the present invention also provides a blood cell composition obtained by inducing differentiation of hematopoietic progenitor cells and having a high content of megakaryocytes.
- the “blood cell composition” includes “megakaryocytes” produced by the method of the present invention, megakaryocytes prepared by other methods, or other blood cells. It may be done.
- hematopoietic progenitor cells are treated by the method of the present invention, differentiation into megakaryocytes can be promoted. Therefore, for example, by applying the method of the present invention to hematopoietic progenitor cells differentiated from pluripotent stem cells or the like, a cell composition having a high megakaryocyte content can be obtained.
- the content of megakaryocytes in the blood cell composition is high can be determined by those skilled in the art based on experience and literature.
- the content of megakaryocytes is at least 20% or more, 30% or more, preferably 40% or more, 50%, compared with the case of treating by other methods. % Or more, more preferably 80% or more. Therefore, according to the method of the present invention, it is possible to prepare a megakaryocyte population or a blood cell population having a high ratio of megakaryocytes.
- Megakaryocytes and the like obtained by the method of the present invention are also effective for transplanting in vivo and producing functional platelets in vivo by an appropriate method. Therefore, the therapeutic agent containing the megakaryocyte obtained by the method of this invention is provided.
- the transplantation of megakaryocytes and the like obtained by the method of the present invention can solve the problem of insufficient donor number and donor burden in bone marrow transplantation and the problem of platelet production ability in vivo in cord blood transplantation. Compared to the transplantation method, it can be said to be a very excellent method.
- the method for producing platelets according to the present invention is to produce platelets in vitro from megakaryocytes obtained by the method of the present invention.
- the method for producing platelets according to the present invention includes a step of culturing megakaryocytes obtained by the above-described method and collecting platelets from the culture.
- the culture conditions are not limited. For example, in the presence of TPO (10 to 200 ng / mL, preferably about 50 to 100 ng / mL), or TPO (10 to 200 ng / mL, preferably 50 to 100 ng / mL). Degree), SCF (10 to 200 ng / mL, preferably about 50 ng / mL) and Heparin (10 to 100 U / mL, preferably about 25 U / ml) may be cultured. The culture can be continued as long as the platelet function is maintained. For example, the culture period is about 7 to 15 days.
- the culture temperature is not particularly limited as long as the effect of the present invention can be obtained, and can be performed at 35 ° C. to 40 ° C., but 37 ° C. to 39 ° C. is preferable.
- the step of culturing megakaryocytes may be performed under serum-free and / or feeder cell-free conditions.
- the method is carried out by culturing megakaryocytes produced according to the method of the present invention in a medium containing TPO.
- a medium containing TPO In the platelet production process, if it can be performed free of serum and feeder cells, the problem of immunogenicity hardly occurs when the obtained platelet is used clinically.
- platelets can be produced without using feeder cells, it is not necessary to adhere feeder cells, so suspension culture can be performed in a flask or the like, which can suppress manufacturing costs and is suitable for mass production.
- conditioned medium may be used.
- the conditioned medium is not particularly limited and can be produced by a person skilled in the art according to a known method.
- the conditioned medium can be obtained by appropriately culturing feeder cells and removing the feeder cells from the culture with a filter.
- a ROCK inhibitor and / or an actomyosin complex function inhibitor is added to the medium.
- a ROCK inhibitor and an actomyosin complex function inhibitor the same thing as what was used by the manufacturing method of the multinucleated megakaryocyte mentioned above can be used.
- ROCK inhibitors include Y27632.
- the actomyosin complex function inhibitor include blebbistatin, which is a myosin heavy chain II ATPase inhibitor.
- a ROCK inhibitor may be added alone, a ROCK inhibitor and an actomyosin complex function inhibitor may be added alone, or a combination thereof may be added.
- the ROCK inhibitor and / or the actomyosin complex function inhibitor is preferably added at 0.1 ⁇ M to 30 ⁇ M, for example, 0.5 ⁇ M to 25 ⁇ M, 5 ⁇ M to 20 ⁇ M, and the like.
- the culture period after adding the ROCK inhibitor and / or the actomyosin complex function inhibitor may be 1 to 15 days, and may be 3 days, 5 days, 7 days, or the like.
- the platelets obtained in the present invention can be administered to patients as a preparation.
- platelets obtained by the method of the present invention are, for example, human plasma, infusion solution, citrate-containing physiological saline, a solution containing glucose-added acetate Ringer solution, PAS (platelet additive solution) (Gulliksson, H. et al., Transfusion, 32: 435-440, (1992)), etc.
- the storage period is about 3 to 7 days, preferably 4 days. As storage conditions, it is desirable to store with shaking and stirring at room temperature (20-24 degrees).
- kits for producing megakaryocytes and / or platelets include kits for producing megakaryocytes and / or platelets.
- the kit includes an apoptosis-suppressing gene, an oncogene, the expression vectors and reagents necessary for expressing the genes (i) to (iii) in cells, a medium for cell culture, serum , Supplements such as growth factors (eg, TPO, EPO, SCF, Heparin, IL-6, IL-11, etc.), antibiotics and the like.
- growth factors eg, TPO, EPO, SCF, Heparin, IL-6, IL-11, etc.
- kits for measuring the expression of KLF1 and / or FLI1 may be included. Reagents, antibodies, and the like contained in the kit are supplied into any type of container in which the constituents are kept active for a long period of time, are not adsorbed by the material of the container, and do not undergo alteration.
- the origin of the “cell” described in the present specification is a human or non-human animal (eg, mouse, rat, cow, horse, pig, sheep, monkey, dog, cat, bird, etc.) and is not particularly limited. But. Particularly preferred are human-derived cells.
- the present invention will be described in more detail with reference to examples, but the present invention is not limited to the examples.
- TKDN SeV2 human fetal skin fibroblast-derived iPS cells established using Sendai virus
- 585A1 585B1, 606A1, 648B1 and 692D2 human peripheral blood mononuclear cell-derived iPS cells established using episomal vectors described in Okita K, et al, Stem Cells 31, 458-66, 2012), Takayama N ., et al. J Exp Med. 2817-2830 (2010), differentiation culture into blood cells was performed.
- human ES / iPS cell colonies were co-cultured with C3H10T1 / 2 feeder cells in the presence of 20 ng / mL VEGF (R & D SYSTEMS) for 14 days to prepare hematopoietic progenitor cells (HPC).
- the culture conditions were 20% O 2 and 5% CO 2 (the same conditions unless otherwise specified).
- Virus infection of hematopoietic progenitor cells On the 6-well plate previously seeded with C3H10T1 / 2 feeder cells, seed HPC obtained by the above method at 5x10 4 cells / well and c-Myc by lentivirus method. And BCL-xL were forcibly expressed. At this time, 6 wells were used for each type of cell line. That is, virus particles were added to the culture medium so as to have an MOI of 20, respectively, and infection was performed by spin infection (32 ° C, 900 rpm, centrifugation for 60 minutes). This operation was performed twice every 12 hours.
- the basic medium (15% Fetal Bovine Serum (GIBCO), 1% Penicillin-Streptomycin-Glutamine (GIBCO), 1% Insulin, Transferrin, Selenium Solution (ITS-G) (GIBCO), 0.45 mM 1-Thioglycerol (Sigma) -Aldrich), IMDM (Iscove's Modified Dulbecco's Medium) (Sigma-Aldrich)) containing 50 ⁇ g / mL L-Ascorbic Acid (Sigma-Aldrich)) 50 ng / mL Human thrombopoietin (TPO) (R & D SYSTEMS), 50 ng / ml Human A medium (hereinafter referred to as differentiation medium) containing Stem Cell Factor (SCF) (R & D SYSTEMS) and 2 ⁇ g / mL Doxycyclin (Dox) was used.
- GEBCO Fetal Bovine Serum
- ITS-G Insulin
- the lentiviral vector is an inducible vector controlled by Tetracycline, and the mOKS cassette of LV-TRE-mOKS-Ubc-tTA-I2G (Kobayashi, T., et al. Cell 142, 787-799 (2010)) Produced by recombination with Bcl-xL and c-Myc (LV-TRE-BCL-xL-Ubc-tTA-I2G and LV-TRE-c-Myc-Ubc-tTA-I2G, respectively).
- the virus particles used for infection were prepared by expressing the above lentiviral vector in 293T cells.
- Infection day 12 Passage. The same operation as on the 6th day of infection was performed. After counting the number of cells, the cells were seeded in 3 ⁇ 10 5 cells / 10 mL / 100 mm dish. ⁇ Infection day 18: passage. The same operation as on the 6th day of infection was performed. After counting the number of cells, the cells were seeded in 3 ⁇ 10 5 cells / 10 mL / 100 mm dish. -Day 24 of infection: passage, cryopreservation, FACS analysis. A part of the cells was passaged (1 ⁇ 10 5 cells / well) in the same manner as described above, and the rest was stored frozen (about 5 ⁇ 10 5 cells / tube). Thereafter, subculture was performed every 4-7 days, and maintenance culture was performed. The medium was not changed during that time.
- the determination of the megakaryocyte strain was determined by the following method. Blood cells were collected on the 24th day of infection, and 2 ⁇ L each of anti-human CD41a-APC antibody (BioLegend), anti-human CD42b-PE antibody (eBioscience), and anti-human CD235ab-pacific blue antibody per 1.0 ⁇ 10 5 cells, The immunostaining was performed using 1 ⁇ L and 1 ⁇ L each and then analyzed using a FACSAria TM II cell sorter (BD) to confirm the establishment of a megakaryocyte strain. Furthermore, normal iPS cell-derived megakaryocytes have a decreased number of cells after 10 days in this differentiation system (Takayama N., et al. J Exp Med. 2817-2830 (2010)). The establishment of a megakaryocyte strain was confirmed by the continued proliferation of CD41a + cells on the day.
- BD FACSAria TM II cell sorter
- megakaryocyte strains were similarly established using hematopoietic progenitor cells derived from iPS cells (TKDN SeV2), and wells continuously growing in infected cells were selected and the number of cells was counted (FIG. 1A). It was confirmed that the cell growth rate was slightly low in the strain with many adherent cells. In addition, it was confirmed that it can be passaged for at least 40 days.
- TKDN SeV2 hematopoietic progenitor cells derived from iPS cells
- the gene expression analysis was performed using a universal probe or a taqman probe after RNA extraction and cDNA conversion according to a conventional method.
- the analyzed genes are GAPDH, c-Myc, Bcl-xL, GATA1, p45 NF-E2, beta1-tubulin, and c-MPL.
- Retroviral vectors are pMXs retro vectors (see Takahashi K, et al, Cell .; 131: 861-872, 2007 or Ohmine K, et al, Oncogene 20, 8249-8257, 2001), pGCDNsam retro vector (Chiba) And received from Professor Iwama University).
- the obtained megakaryocyte progenitor cells showed a basophilic monoblast-like morphology (FIG. 7B), and produced CD41a-positive abnormal platelet-like particles with slightly low CD42b expression. This seems to be because the forced expression of c-Myc was maintained.
- a vector expressing c-MYC having an unstable domain (DD (Destabilization Domain)) at the C-terminus was used.
- DD Destabilization Domain
- a vector expressing c-MYC-DD-2A-BMI1 was constructed using pPTunerC vector and Shied-1 (Clontech / Takara Bio).
- CD41a-positive megakaryocyte progenitor cells could be expanded for at least 50 days (FIG. 8A).
- expansion culture could not be maintained with c-MYC-2A-BMI1.
- megakaryocyte progenitor cells were induced using four iPS cell clones using the following two protocols (Cl-3: derived from KhES3 strain, Cl-4: 692D2). Strain-derived, Cl-6: 585A1 strain and Cl-7: TKDN SeV2 strain); (1) Method of introducing BCL-XL, c-MYC and BMI1 simultaneously, and (2) BCL-XL and c-MYC After 14 days, the method will be introduced 14 to 21 days later.
- megakaryocyte progenitor cells could be expanded for up to 40 to 50 days in any iPS cell clone, but BCL-XL was expressed later.
- expansion culture could be continued for 60 days or longer when any ES cell or iPS cell clone was used (FIGS. 10B, C, D and E).
- FIG. 12A shows a mature example that can be confirmed by strong expression of CD42b in megakaryocyte progenitor cells (Cl-2 and Cl-7) derived from two clones on the fourth day after expression cessation. It was confirmed that the expression of GATA1, FOG1, NF-E2, and ⁇ 1-tubulin was enhanced by maturation of megakaryocyte progenitor cells into megakaryocytes.
- Platelets were obtained, and Cl-7 yielded 10 platelets. Similarly, the expression of foreign genes was stopped from megakaryocyte progenitor cells using 10 cm dishes (10 ml of medium) to produce platelets, and 4 ⁇ 10 6 (Cl-7) or 2 ⁇ 10 6 per 1 ml of medium was produced. Six (Cl-2) platelets were confirmed (FIG. 13D). This suggests that in order to obtain 10 11 platelets necessary for a single platelet transfusion, it can be produced by culturing megakaryocyte progenitor cells in a 25-50 L medium.
- imMKCL platelets Platelets obtained by the above method (after stopping expression of foreign genes and cultured in serum-free medium for 5 days) (hereinafter referred to as imMKCL platelets) were observed with a scanning electron microscope. Compared with human platelets (hereinafter referred to as “Fresh platelet”), there were slightly fewer granules (FIG. 14A).
- imMKCL platelets or Fresh platelets were mixed with the same number of Fresh platelets, 100 ⁇ M ADP and 100 ⁇ M TRAP6, or 10 ⁇ g / mL collagen (Nycomed) was added, and the mixture was stimulated at 37 ° C. for 10 minutes under shaking.
- platelet aggregation was also confirmed in imMKCL platelets (FIGS. 14D and E).
- a clot test (coagulation test) stimulated with 2 U / ml thrombin was added to IMDM containing 20% platelet-free plasma and coagulated in response to thrombin stimulation. was confirmed.
- imMKCL platelets suggested initial adhesion to the blood vessel wall depending on P-selectin.
- imMKCL platelets prepared from four iPS cell clones were tested in the same manner, it was confirmed that they could be more involved in thrombus formation than pooled platelets (FIGS. 15E and F).
Abstract
Description
[1] 以下の(i)~(ii)の工程を含む、造血前駆細胞から巨核球を製造する方法;
(i)アポトーシス抑制遺伝子および癌遺伝子を造血前駆細胞において強制発現させて培養する工程、および
(ii)工程(i)で得られた細胞について、アポトーシス抑制遺伝子および癌遺伝子の強制発現を止めて培養する工程。
[2] 前記工程(i)において、p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子をさらに造血前駆細胞において強制発現させ、前記工程(ii)において、当該p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子の強制発現を止めて培養する、[1]に記載の方法。
[3] 前記工程(i)が、癌遺伝子、ならびにp16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子を造血前駆細胞において強制発現させた後、アポトーシス抑制遺伝子をさらに当該細胞へ強制発現させる工程である、[2]に記載の方法。
[4] 前記工程(i)において、癌遺伝子、ならびにp16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子を造血前駆細胞において少なくとも28日強制発現させて培養した後、アポトーシス抑制遺伝子をさらに当該細胞へ強制発現させる工程である、[3]に記載の方法。
[5] 前記アポトーシス抑制遺伝子が、BCL-XL遺伝子である、[1]から[4]のいずれか1項に記載の方法。
[6] 前記癌遺伝子が、c-MYC遺伝子である、[1]から[5]のいずれか1項に記載の方法。
[7] 前記p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子が、BMI1である、[1]から[6]のいずれか1項に記載の方法。
[8] 前記工程(i)および(ii)において、TPOを含有する培養液中でC3H10T1/2細胞上で該細胞を培養する、[1]から[7]のいずれか1項に記載の方法。
[9] 前記工程(i)および(ii)での培養において、SCFをさらに含有する培養液中で培養する、[8]に記載の方法。
[10] 前記遺伝子の強制発現が、薬剤応答性ベクターを用いて行われる、[1]から[9]のいずれか1項に記載の方法。
[11] 前記造血前駆細胞が、多能性幹細胞から分化誘導された細胞である、[1]から[10]のいずれか1項に記載の方法。
[12] 前記造血前駆細胞が、多能性幹細胞から分化誘導された細胞である、[11]に記載の方法であって、該分化誘導において、多能性幹細胞をVEGFを含有する培養液中でC3H10T1/2細胞上で培養する工程を含む、方法。
[13] 前記造血前駆細胞において、KLF1の発現が低い、またはFLI1の発現が高い、[1]から[12]のいずれか1項に記載の方法。
[14] 前記造血前駆細胞におけるKLF1またはFLI1の発現が、それぞれKhES3由来の造血前駆細胞での発現と比較してより低いまたはより高い、[13]に記載の方法。
[15] 工程(i)に先立って、前記造血前駆細胞におけるKLF1および/またはFLI1の発現を測定する工程を含む、[1]から[14]のいずれか1項に記載の方法。
[16] 前記工程(ii)を、5日間行う、[1]から[15]のいずれか1項に記載の方法。
[17] 血小板の製造方法であって、[1]から[16]のいずれか1項に記載の方法で得られた巨核球の培養物から血小板を回収する工程を含む方法。
[18] [17]に記載の方法で製造された血小板。
[19] [18]に記載の血小板を含む血液製剤。
[20] 以下の(I)~(II)の工程を含む、造血前駆細胞から巨核球前駆細胞を製造する方法;
(I)癌遺伝子およびp16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子を造血前駆細胞において強制発現させて培養する工程、および
(II)工程(I)で得られた細胞へさらにアポトーシス抑制遺伝子を強制発現させる、またはカスパーゼ阻害剤を添加した培地で培養する工程。
[21] 前記アポトーシス抑制遺伝子が、BCL-XL遺伝子である、[20]に記載の方法。
[22] 前記癌遺伝子が、c-MYC遺伝子である、[20]から[21]のいずれか1項に記載の方法。
[23] 前記カスパーゼ阻害剤が、Z-DEVD-FMKである、[20]から[22]のいずれか1項に記載の方法。
[24] 前記p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子が、BMI1である、[20]から[23]のいずれか1項に記載の方法。
[25] 前記工程(I)を、少なくとも28日間行う、[20]から[24]のいずれか1項に記載の方法。
[26] 前記巨核球前駆細胞が拡大培養可能な細胞である、[20]から[25]のいずれか1項に記載の方法。
[27] 薬剤応答性で発現する外来性のアポトーシス抑制遺伝子および癌遺伝子が染色体に組み込まれている巨核球であって、当該外来性の遺伝子が発現していない細胞。
[28] 薬剤応答性で発現する外来性のp16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子がさらに染色体に組み込まれており、当該外来性遺伝子が発現していない、[27]に記載の細胞。
[29] 前記アポトーシス抑制遺伝子が、BCL-XL遺伝子である、[27]または[28]に記載の細胞。
[30] 前記癌遺伝子が、c-MYC遺伝子である、[27]から[29]のいずれか1項に記載の細胞。
[31] 前記p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子が、BMI1である、[27]から[30]のいずれか1項に記載の細胞。
[32] 巨核球の製造に適した造血前駆細胞を選択する方法であって、KLF1の発現またはFLI1の発現を測定する工程を含む方法。
[33] 前記KLF1の発現が低い造血前駆細胞を選択する工程を含む、[32]に記載の方法。
[34] 前記FLI1の発現が高い造血前駆細胞を選択する工程を含む、[33]に記載の方法。
[35] 前記造血前駆細胞が、多能性幹細胞から分化誘導された細胞である、[32]から[33]のいずれか1項に記載の方法。
[36] 以下の工程を含む巨核球製造に適した多能性幹細胞を選択する方法;
(i)多能性幹細胞から造血幹細胞を製造する工程、および、
(ii)工程(i)で製造された造血前駆細胞において、KLF1の発現およびFLI1の発現の測定する工程。
に関する。
また、本発明によれば、巨核球を製造するために適した造血前駆細胞を選択することが可能となる。
本発明は、造血前駆細胞から巨核球を製造する方法を提供する。
本発明に係る巨核球の製造方法の一態様は、造血前駆細胞において、アポトーシス抑制遺伝子および癌遺伝子を強制発現させて該細胞を培養する工程およびアポトーシス抑制遺伝子および癌遺伝子の強制発現を止めて培養する工程を含む。本発明において、造血前駆細胞において、アポトーシス抑制遺伝子および癌遺伝子を強制発現させて該細胞を培養する工程によって得られる細胞を巨核球前駆細胞としても良い。
ES細胞は、ヒトやマウスなどの哺乳動物の初期胚(例えば胚盤胞)の内部細胞塊から樹立された、多能性と自己複製による増殖能を有する幹細胞である。
ES細胞は、受精卵の8細胞期、桑実胚後の胚である胚盤胞の内部細胞塊に由来する胚由来の幹細胞であり、成体を構成するあらゆる細胞に分化する能力、いわゆる分化多能性と、自己複製による増殖能とを有している。ES細胞は、マウスで1981年に発見され (M.J. Evans and M.H. Kaufman (1981), Nature 292:154-156)、その後、ヒト、サルなどの霊長類でもES細胞株が樹立された (J.A. Thomson et al. (1998), Science 282:1145-1147; J.A. Thomson et al. (1995), Proc. Natl. Acad. Sci. USA, 92:7844-7848;J.A. Thomson et al. (1996), Biol. Reprod., 55:254-259; J.A. Thomson and V.S. Marshall (1998), Curr. Top. Dev. Biol., 38:133-165)。
精子幹細胞は、精巣由来の多能性幹細胞であり、精子形成のための起源となる細胞である。この細胞は、ES細胞と同様に、種々の系列の細胞に分化誘導可能であり、例えばマウス胚盤胞に移植するとキメラマウスを作出できるなどの性質をもつ(M. Kanatsu-Shinohara et al. (2003) Biol. Reprod., 69:612-616; K. Shinohara et al. (2004), Cell, 119:1001-1012)。神経膠細胞系由来神経栄養因子(glial cell line-derived neurotrophic factor (GDNF))を含む培養液で自己複製可能であるし、またES細胞と同様の培養条件下で継代を繰り返すことによって、精子幹細胞を得ることができる(竹林正則ら(2008),実験医学,26巻,5号(増刊),41~46頁,羊土社(東京、日本))。
胚性生殖細胞は、胎生期の始原生殖細胞から樹立される、ES細胞と同様な多能性をもつ細胞であり、LIF、bFGF、幹細胞因子(stem cell factor)などの物質の存在下で始原生殖細胞を培養することによって樹立しうる(Y. Matsui et al. (1992), Cell, 70:841-847; J.L. Resnick et al. (1992), Nature, 359:550-551)。
人工多能性幹(iPS)細胞は、特定の初期化因子を、DNA又はタンパク質の形態で体細胞に導入することによって作製することができる、ES細胞とほぼ同等の特性、例えば分化多能性と自己複製による増殖能、を有する体細胞由来の人工の幹細胞である(K. Takahashi and S. Yamanaka (2006) Cell, 126:663-676; K. Takahashi et al. (2007), Cell, 131:861-872; J. Yu et al. (2007), Science, 318:1917-1920; Nakagawa, M.ら,Nat. Biotechnol. 26:101-106 (2008);国際公開WO 2007/069666)。初期化因子は、ES細胞に特異的に発現している遺伝子、その遺伝子産物もしくはnon-cording RNAまたはES細胞の未分化維持に重要な役割を果たす遺伝子、その遺伝子産物もしくはnon-coding RNA、あるいは低分子化合物によって構成されてもよい。初期化因子に含まれる遺伝子として、例えば、Oct3/4、Sox2、Sox1、Sox3、Sox15、Sox17、Klf4、Klf2、c-Myc、N-Myc、L-Myc、Nanog、Lin28、Fbx15、ERas、ECAT15-2、Tcl1、beta-catenin、Lin28b、Sall1、Sall4、Esrrb、Nr5a2、Tbx3またはGlis1等が例示され、これらの初期化因子は、単独で用いても良く、組み合わせて用いても良い。初期化因子の組み合わせとしては、WO2007/069666、WO2008/118820、WO2009/007852、WO2009/032194、WO2009/058413、WO2009/057831、WO2009/075119、WO2009/079007、WO2009/091659、WO2009/101084、WO2009/101407、WO2009/102983、WO2009/114949、WO2009/117439、WO2009/126250、WO2009/126251、WO2009/126655、WO2009/157593、WO2010/009015、WO2010/033906、WO2010/033920、WO2010/042800、WO2010/050626、WO 2010/056831、WO2010/068955、WO2010/098419、WO2010/102267、WO 2010/111409、WO 2010/111422、WO2010/115050、WO2010/124290、WO2010/147395、WO2010/147612、Huangfu D, et al. (2008), Nat. Biotechnol., 26: 795-797、Shi Y, et al. (2008), Cell Stem Cell, 2: 525-528、Eminli S, et al. (2008), Stem Cells. 26:2467-2474、Huangfu D, et al. (2008), Nat Biotechnol. 26:1269-1275、Shi Y, et al. (2008), Cell Stem Cell, 3, 568-574、Zhao Y, et al. (2008), Cell Stem Cell, 3:475-479、Marson A, (2008), Cell Stem Cell, 3, 132-135、Feng B, et al. (2009), Nat Cell Biol. 11:197-203、R.L. Judson et al., (2009), Nat. Biotech., 27:459-461、Lyssiotis CA, et al. (2009), Proc Natl Acad Sci U S A. 106:8912-8917、Kim JB, et al. (2009), Nature. 461:649-643、Ichida JK, et al. (2009), Cell Stem Cell. 5:491-503、Heng JC, et al. (2010), Cell Stem Cell. 6:167-74、Han J, et al. (2010), Nature. 463:1096-100、Mali P, et al. (2010), Stem Cells. 28:713-720、Maekawa M, et al. (2011), Nature. 474:225-9.に記載の組み合わせが例示される。
nt ES細胞は、核移植技術によって作製されたクローン胚由来のES細胞であり、受精卵由来のES細胞とほぼ同じ特性を有している(T. Wakayama et al. (2001), Science, 292:740-743; S. Wakayama et al. (2005), Biol. Reprod., 72:932-936; J. Byrne et al. (2007), Nature, 450:497-502)。すなわち、未受精卵の核を体細胞の核と置換することによって得られたクローン胚由来の胚盤胞の内部細胞塊から樹立されたES細胞がnt ES(nuclear transfer ES)細胞である。nt ES細胞の作製のためには、核移植技術(J.B. Cibelli et al. (1998), Nature Biotechnol., 16:642-646)とES細胞作製技術との組み合わせが利用される(若山清香ら(2008),実験医学,26巻,5号(増刊), 47~52頁)。核移植においては、哺乳動物の除核した未受精卵に、体細胞の核を注入し、数時間培養することで初期化することができる。
Muse細胞は、WO2011/007900に記載された方法にて製造された多能性幹細胞であり、詳細には、線維芽細胞または骨髄間質細胞を長時間トリプシン処理、好ましくは8時間または16時間トリプシン処理した後、浮遊培養することで得られる多能性を有した細胞であり、SSEA-3およびCD105が陽性である。
STAP細胞は、WO2013/163296に記載された方法にて製造された多能性幹細胞であり、例えば、体細胞をpH5.4から5.8の酸性溶液中で30分間培養して得られる、SSEA-4およびE-cadherinが陽性の細胞である。
(i)p16遺伝子又はp19遺伝子の発現を抑制する遺伝子;
(ii)Ink4a/Arf遺伝子の発現を抑制する遺伝子;及び
(iii)ポリコーム遺伝子。
(1)Z-Asp-CH2-DCB(分子量454.26)
(2)Boc-Asp(OMe)-FMK(分子量263.3)
(3)Boc-Asp(OBzl)-CMK(分子量355.8)
(4)Ac-AAVALLPAVLLALLAP-YVAD-CHO(分子量1990.5)(配列番号:1)
(5)Ac-AAVALLPAVLLALLAP-DEVD-CHO(分子量2000.4)(配列番号:2)
(6)Ac-AAVALLPAVLLALLAP-LEVD-CHO(分子量1998.5)(配列番号:3)
(7)Ac-AAVALLPAVLLALLAP-IETD-CHO(分子量2000.5)(配列番号:4)
(8)Ac-AAVALLPAVLLALLAP-LEHD-CHO(分子量2036.5)(配列番号:5)
(9)Z-DEVD-FMK (Z-Asp-Glu-Val-Asp-fluoromethylketone)(配列番号:6)
(10)Z-VAD FMK
例えば、ペプチド性化合物のカスパーゼ阻害剤として、(1)VX-740 - Vertex Pharmaceuticals (Leung-Toung et al., Curr. Med. Chem. 9, 979-1002 (2002))、(2)HMR-3480 - Aventis Pharma AG (Randle et al., Expert Opin. Investig. Drugs 10, 1207-1209 (2001))、を挙げることができる。
本発明はまた、造血前駆細胞を分化誘導して得られる血液細胞組成物であって、巨核球の含有率が高い血液細胞組成物を提供する。ここで、「血液細胞組成物」には、本発明の方法で製造された「巨核球」を含む他、その他の方法によって調製された巨核球、又は、その他の血液細胞が含まれて又は加えられていてもよい。
本発明の方法によって造血前駆細胞を処理すると、巨核球への分化を促進することができる。そのため、例えば、多能性幹細胞などから分化させた造血前駆細胞に対し、本発明の方法を適用することで、巨核球の含有率の高い細胞組成物を取得することができる。血液細胞組成物において巨核球の含有率が高いか否かは、当業者が経験や文献に基づいて判断することができる。本発明の方法で造血前駆細胞を処理した場合、他の方法で処理した場合と比較して、巨核球の含有率を、少なくとも、20%以上、30%以上、好ましくは、40%以上、50%以上、より好ましくは80%以上にまで増大させることが可能である。従って、本発明の方法により、巨核球の存在比率の高い巨核球集団、あるいは、血液細胞集団を調製することが可能となる。
本発明の方法によって得られる巨核球等の移植は、骨髄移植におけるドナー数の不足及びドナーの負担の問題、臍帯血移植における生体内での血小板産生能力の問題を解決することが可能で、従来の移植法と比較して、非常に優れた方法ということができる。
本発明に係る血小板の製造方法は、本発明の方法で得られた巨核球から、in vitroで血小板を産生させるものである。
本発明の実施形態には、巨核球及び/または血小板を製造するためのキットが含まれる。当該キットには、アポトーシス抑制遺伝子、癌遺伝子、上記(i)~(iii)の遺伝子等を細胞内で発現するのに必要な発現ベクター等及び試薬などの他、細胞培養のための培地、血清、増殖因子などのサプリメント(例えば、TPO、EPO、SCF、Heparin、IL-6、IL-11など)、抗生物質などが含まれる。その他、例えば、多能性細胞由来の細胞を使用する場合、これらの細胞から調製したネット様構造物を同定するためのマーカー確認用の抗体(例えば、Flk1、CD31、CD34、UEA-Iレクチンなどに対する抗体)なども含まれる。さらに、巨核球の製造に適した造血前駆細胞を選択するため、KLF1及び/またはFLI1の発現を測定するキットを含んでも良い。キット中に含まれる試薬、抗体等は、構成成分が活性を長期間有効に持続し、容器の材質によって吸着されず、変質を受けないような何れかの種類の容器中に供給される。
以下に実施例を示してさらに詳細に説明するが、本発明は実施例により何ら限定されるものではない。
ヒトES細胞(khES3:京都大学より入手)およびiPS細胞(TKDN SeV2:センダイウィルスを用いて樹立されたヒト胎児皮膚繊維芽細胞由来iPS細胞、585A1、585B1、606A1、648B1および692D2:Okita K, et al, Stem Cells 31, 458-66, 2012に記載のエピソーマルベクターを用いて樹立されたヒト末梢血単核球由来iPS細胞)から、Takayama N., et al. J Exp Med. 2817-2830 (2010)に記載の方法に従って、血球細胞への分化培養を実施した。即ち、ヒトES/iPS細胞コロニーを20ng/mL VEGF (R&D SYSTEMS)存在下でC3H10T1/2フィーダー細胞と14日間共培養して造血前駆細胞(Hematopoietic Progenitor Cells(HPC)) を作製した。培養条件は20% O2、5% CO2で実施した(特に記載がない限り、以下同条件)。
予めC3H10T1/2フィーダー細胞を播種した6 well plate上に、上記の方法で得られたHPCを5x104cells/wellずつ播種し、レンチウィルス法にてc-MycおよびBCL-xLを強制発現させた。このとき、細胞株1種類につき6 wellずつ使用した。即ち、それぞれMOI 20になるように培地中にウィルス粒子を添加し、スピンインフェクション(32℃ 900rpm, 60分間遠心)で感染させた。本操作は、12時間おきに2回実施した。このとき、基本培地(15% Fetal Bovine Serum (GIBCO)、1% Penicillin-Streptomycin-Glutamine (GIBCO)、1% Insulin, Transferrin, Selenium Solution (ITS -G) (GIBCO)、0.45mM 1-Thioglycerol (Sigma-Aldrich)、50μg/mL L-Ascorbic Acid (Sigma-Aldrich)を含有するIMDM (Iscove’s Modified Dulbecco’s Medium) (Sigma-Aldrich))へ50ng/mL Human thrombopoietin (TPO) (R&D SYSTEMS)、50ng/ml Human Stem Cell Factor (SCF) (R&D SYSTEMS)および2μg/mL Doxycyclin (Dox)を加えた培地(以下、分化培地)を用いた。なお、レンチウィルスベクターは、Tetracycline制御性のinducible vectorであり、LV-TRE-mOKS-Ubc-tTA-I2G(Kobayashi, T., et al. Cell 142, 787-799 (2010))のmOKSカセットをBcl-xLおよびc-Mycに組み替えることで作製された(それぞれ、LV-TRE-BCL-xL-Ubc-tTA-I2GおよびLV-TRE- c-Myc-Ubc-tTA-I2G)。感染に用いたウィルス粒子は、293T細胞へ上記レンチウィルスベクターを発現させて作製された。
上記の方法でウィルス感染を実施した日を感染0日目として、以下の通り、巨核球株を培養し、樹立した。
・感染2日目:継代。
ピペッティングにて上記の方法で得られたウィルス感染した細胞を回収し、1200rpm, 5分間遠心操作を行って上清を除去した後、新しい分化培地で懸濁して新しいC3H10T1/2フィーダー細胞上に播種した(6well plate)。
・感染6日目:継代。
感染2日目と同様の操作を実施。ただし、樹立は複数回行い、このうち1回目(Exp.1)ではSCFを添加しない分化培地でおこなった。SCFを添加した方が増殖がよい印象を得られた。
・感染12日目:継代。
感染6日目と同様の操作を実施。細胞数を計測後3×105cells/ 10mL / 100 mm dishで播種した。
・感染18日目:継代。
感染6日目と同様の操作を実施。細胞数を計測後3×105cells/ 10mL / 100 mm dishで播種した。
・感染24日目:継代、凍結保存、FACS解析。
細胞の一部を上記と同様に継代(1×105cells/well)し、残りを凍結保存した(5×105 cells/tube程度)。以後、4-7日毎に継代を行い、維持培養を行った。その間の培地替えは行わなかった。
ES細胞(khES3)およびiPS細胞(TKDN SeV2)由来の造血前駆細胞から上述の方法で巨核球株の樹立を試みたところ、TKDN SeV2由来の造血前駆細胞からは6例中3例にて巨核球株の樹立が確認されたが、KhES3由来では巨核球株が6例中では樹立できなかった(図1A)。
感染24日目に巨核球株を5.0×105cells/wellずつ、C3H10T1/2フィーダー細胞上でDox含有あるいは不含の分化培地(SCFについては添加および非添加の2条件にて行った)を用いて3あるいは5日間培養した。それぞれ導入遺伝子発現条件(Gene-ON)、導入遺伝子停止条件(Gene-OFF)とする。ピペッティングで培養液を回収し、細胞の増殖速度(図2)、血球分画および血小板分画のFACS解析(図3および4)および遺伝子発現解析(図5)に供した。FACS解析は上述した方法と同様に実施した。遺伝子発現解析は定法に従い、RNA抽出し、cDNA化を行った後、universal probeあるいはtaqman probeを用いて実施した。解析した遺伝子はGAPDH、c-Myc、Bcl-xL、GATA1、p45 NF-E2、beta1-tubulin、c-MPLである。
上記の方法でiPS細胞(TKDN SeV2)から作製した巨核球株(培養40日目)およびTakayamaら,Blood,111:5298-5306 2008に記載の方法でES細胞(khES3)から作製した巨核球(分化誘導開始から21日目)に対して、Phorbol 12-Myristate 13-acetate (PMA)刺激した直後のFibrinogenへの結合能を測定した(図6)。その結果、本発明の方法を用いて作製した巨核球株では、PMA刺激に反応してFibrinogenへの結合能を有することが確認できたが、従来の方法で得られた巨核球ではPMA刺激後においても顕著なFibrinogen結合能は有さなかった。以上より、本発明の方法で作製された巨核球では、より成熟した巨核球が作製できることが示唆された。
実施例1に記載の方法で得られたKhES3由来のHPCへ(1)c-Mycのみ、(2)Bmi1のみ、(3)c-MYCおよびsh-p53、(4)c-MYCおよびBCL-XL、(5)c-MYCおよびsh-ARF、(6)c-MYCおよびBMI1、または(7)c-MYC、sh-INK4Aおよびsh-ARFを各遺伝子に1つのレトロウィルスベクターを用いて導入し、基本培地へ50ng/mL TPOおよび50ng/ml SCFを加えた培地で培養したところ、少なくともc-MYCを導入した場合は、CD41a、CD42a、CD42bおよびCD9が陽性である巨核球前駆細胞が得られた。さらに培養を継続したところ、(6)c-MYCおよびBMI1、ならびに(7)c-MYC、sh-INK4Aおよびsh-ARFについては、2ヶ月間につづき拡大培養することが可能であった(図7A)。レトロウィルスベクターはpMXsレトロベクター(Takahashi K, et al, Cell.;131:861-872, 2007またはOhmine K, et al, Oncogene 20, 8249-8257, 2001を参照のこと)、pGCDNsamレトロベクター(千葉大学岩間教授より受領)を用いて導入した。sh-p53は、Brummelkamp TR, et al, Science 296, 550-553, 2002を参照し、sh-INK4Aおよびsh-ARFは、Voorhoeve PM and Agami R, Cell 4, 311-319, 2003を参照して作製した。
c-MYCおよびBMI1の発現様式を図7Cに示したコンストラクトを用いてc-MYC-2A-BMI1またはBMI1-2A-c-MYCをレトロウィルスベクターを用いて強制発現させ、上記と同様の培養条件にて巨核球前駆細胞を誘導したところ、c-MYC-2A-BMI1を用いた場合のみ40日以上の拡大培養が可能であった(図7D)。それぞれの導入方法によるc-Mycの発現量を確認したところ、c-MYC-2A-BMI1を用いた場合、BMI1-2A-c-MYCを用いた場合よりもc-MYCの発現が低いことが確認された(図7E)。そこで、c-Mycの発現を抑制する目的で、不安定ドメイン(DD (Destabilization Domain))をC末端に有するc-MYCを発現するベクターを用いた。DDを有する発現ベクターは、pPTunerC vector and Shied-1 (Clontech/Takara Bio社)を用いて、c-MYC-DD-2A-BMI1を発現するベクターを構築した。このc-MYC-DD-2A-BMI1を上記と同様にHPCへ導入し培養を継続したところ、少なくとも50日間、CD41a陽性の巨核球前駆細胞の拡大培養が可能であった(図8A)。一方、c-MYC -2A-BMI1では、拡大培養を維持することができなかった。このことは、Shield-1を添加し、c-MYCの発現を安定させたところ、容量依存的に巨核球前駆細胞数が減少し、Shield-1そのものの毒性によるものではないことを確認したことから、c-MYCの発現量が拡大培養に影響することが確認された(図8B)。このc-MYCの発現による拡大培養への影響は、Caspase依存型アポトーシスによるものと予想し、Shield-1を添加によるCaspase-3/7の活性を測定したところ、c-MYCの安定化に伴いCaspaseが活性化されることが確認された(図8C)。以上のことから、c-MYCの過剰発現によるアポトーシスのため、巨核球前駆細胞の拡大培養が阻害されていると示唆された。
c-MYCおよびBMI1の強制発現では、巨核球前駆細胞の誘導は可能であるが、c-MYCの発現量に依存したアポトーシスにより拡大培養に限度が生じる。そこで、アポトーシスを抑制するためBCL-XLをc-MYCおよびBMI1の導入後14日から21日後の間に導入したところ(図9A)、iPS細胞由来(Cl-1:692D2株由来)およびES細胞由来(Cl-2:khES3由来)のHPCから誘導した巨核球前駆細胞は、5か月間以上もの拡大培養が可能であることが確認された(図9BおよびC)。さらに、c-MYC-DD、BMI1およびBCL-XLを同時にHPCにて共発現させ、Shield-1の添加量を変えてc-MYCの発現量を調節し、7日目の巨核球細胞数を検討したところ、c-MYCの発現量が高くとも、BCL-XLを発現させることで巨核球前駆細胞が誘導できることが確認された(図9DおよびE)。
上記の通りc-MYCおよびBMI1の導入後、BCL-xLを導入した方法で作製した巨核球前駆細胞株を凍結融解後、同条件で培養したところ、21日間の拡大培養が可能であった(図11A)。このときの細胞マーカーを調べたところ、CD41a、CD42a、CD42bおよびCD9の発現は、凍結前と変化がなかった(図11B)。従って、本方法で製造される巨核球前駆細胞は、凍結保存が可能であることが示された。
上記の方法で得られた巨核球前駆細胞において外来遺伝子であるc-MYC、BMI1およびBCL-XLの発現を、Doxを含有しない培地へ交換することで止めて、5日間培養を続けたところ(図12A)、20.2%の細胞において多核化を示した(図12B)。このときCD42b陽性である血小板前駆体の形成が確認された。さらに、発現停止後4日目における2つのクローン由来の巨核球前駆細胞(Cl-2およびCl-7)のCD42bの強発現化で確認できる成熟例を図12Cに示す。このような巨核球前駆細胞から巨核球への成熟により、GATA1、FOG1、NF-E2およびβ1-tubulinの発現が増強することが確認された。
上記のとおり、外来性のc-MYC、BMI1およびBCL-XLの発現を停止することでCD42bの発現が増強し、CD41a陽性およびCD42b陽性である血小板が得られた(図13A)。いずれの外来遺伝子の発現を停止させることが最も効率よく血小板を産生するのかを検討するため、BCL-XLの発現のみ維持した場合と3つの遺伝子全てを停止した場合を比較したところ、3つの遺伝子全てを停止した場合において最も効率よく血小板が産生されることが確認された(図13B)。さらに既存の巨核球株(Meg01(ATCC)、CMKおよびK562(大阪大学Dr. H. Kashiwagiより受領))を10% FBSおよびPSGを添加したRPMIで培養し、100 nM PMAを添加して生じるCD41a陽性およびCD42b陽性粒子の産生量とiPS細胞由来の巨核球前駆細胞からの同粒子の産生量を比較したところ、iPS細胞由来の巨核球前駆細胞から産生する血小板量が有意に多いことが確認された(図13C)。このとき、既存の巨核球株からは、CD41a陽性およびCD42b陰性の血小板様粒子の数が多く見られた。続いて、外来遺伝子の強制発現を停止して5日間無血清培地において、1つの誘導巨核球前駆細胞から産生されるCD42b陽性の血小板の量を確認したところ、Cl-2では1つの細胞から3個の血小板が得られ、Cl-7では10個の血小板が得られた。同様に、10cmディッシュ(10mlの培地)を用いて巨核球前駆細胞から外来遺伝子の発現を停止させて血小板を産生させたところ、培地1mlあたり4×106個(Cl-7)または2×106個(Cl-2)の血小板が確認された(図13D)。このことから、一度の血小板輸血に必要量である1011個の血小板を得るためには、25~50Lの培地で巨核球前駆細胞を培養することによって製造することができることが示唆された。
2.4Gyの放射線照射後9日目のNOGマウス(血小板減少症モデルマウス)へFresh platelet(1×108)、またはimMKCL血小板(6×108または1×108)を尾静脈より投与して、30分後、2時間後、および24時間後に採血(50から100μL)し、ヒトCD41a陽性の血小板数を測定した(図15A)。その結果、マウス体内におけるヒトCD41a陽性血小板の減少速度は、imMKCL血小板およびFresh plateletにおいて有意差は見られなかった。
Claims (36)
- 以下の(i)~(ii)の工程を含む、造血前駆細胞から巨核球を製造する方法;
(i)アポトーシス抑制遺伝子および癌遺伝子を造血前駆細胞において強制発現させて培養する工程、および
(ii)工程(i)で得られた細胞について、アポトーシス抑制遺伝子および癌遺伝子の強制発現を止めて培養する工程。 - 前記工程(i)において、p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子をさらに造血前駆細胞において強制発現させ、前記工程(ii)において、当該p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子の強制発現を止めて培養する、請求項1に記載の方法。
- 前記工程(i)が、癌遺伝子、ならびにp16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子を造血前駆細胞において強制発現させた後、アポトーシス抑制遺伝子をさらに当該細胞へ強制発現させる工程である、請求項2に記載の方法。
- 前記工程(i)において、癌遺伝子、ならびにp16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子を造血前駆細胞において少なくとも28日強制発現させて培養した後、アポトーシス抑制遺伝子をさらに当該細胞へ強制発現させる工程である、請求項3に記載の方法。
- 前記アポトーシス抑制遺伝子が、BCL-XL遺伝子である、請求項1から4のいずれか1項に記載の方法。
- 前記癌遺伝子が、c-MYC遺伝子である、請求項1から5のいずれか1項に記載の方法。
- 前記p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子が、BMI1である、請求項1から6のいずれか1項に記載の方法。
- 前記工程(i)および(ii)において、TPOを含有する培養液中でC3H10T1/2細胞上で該細胞を培養する、請求項1から7のいずれか1項に記載の方法。
- 前記工程(i)および(ii)での培養において、SCFをさらに含有する培養液中で培養する、請求項8に記載の方法。
- 前記遺伝子の強制発現が、薬剤応答性ベクターを用いて行われる、請求項1から9のいずれか1項に記載の方法。
- 前記造血前駆細胞が、多能性幹細胞から分化誘導された細胞である、請求項1から10のいずれか1項に記載の方法。
- 前記造血前駆細胞が、多能性幹細胞から分化誘導された細胞である、請求項11に記載の方法であって、該分化誘導において、多能性幹細胞をVEGFを含有する培養液中でC3H10T1/2細胞上で培養する工程を含む、方法。
- 前記造血前駆細胞において、KLF1の発現が低い、またはFLI1の発現が高い、請求項1から12のいずれか1項に記載の方法。
- 前記造血前駆細胞におけるKLF1またはFLI1の発現が、それぞれKhES3由来の造血前駆細胞での発現と比較してより低いまたはより高い、請求項13に記載の方法。
- 工程(i)に先立って、前記造血前駆細胞におけるKLF1および/またはFLI1の発現を測定する工程を含む、請求項1から14のいずれか1項に記載の方法。
- 前記工程(ii)を、5日間行う、請求項1から15のいずれか1項に記載の方法。
- 血小板の製造方法であって、請求項1から16のいずれか1項に記載の方法で得られた巨核球の培養物から血小板を回収する工程を含む方法。
- 請求項17に記載の方法で製造された血小板。
- 請求項18に記載の血小板を含む血液製剤。
- 以下の(I)~(II)の工程を含む、造血前駆細胞から巨核球前駆細胞を製造する方法;
(I)癌遺伝子およびp16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子を造血前駆細胞において強制発現させて培養する工程、および
(II)工程(I)で得られた細胞へさらにアポトーシス抑制遺伝子を強制発現させる、またはカスパーゼ阻害剤を添加した培地で培養する工程。 - 前記アポトーシス抑制遺伝子が、BCL-XL遺伝子である、請求項20に記載の方法。
- 前記癌遺伝子が、c-MYC遺伝子である、請求項20から21のいずれか1項に記載の方法。
- 前記カスパーゼ阻害剤が、Z-DEVD-FMKである、請求項20から22のいずれか1項に記載の方法。
- 前記p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子が、BMI1である、請求項20から23のいずれか1項に記載の方法。
- 前記工程(I)を、少なくとも28日間行う、請求項20から24のいずれか1項に記載の方法。
- 前記巨核球前駆細胞が拡大培養可能な細胞である、請求項20から25のいずれか1項に記載の方法。
- 薬剤応答性で発現する外来性のアポトーシス抑制遺伝子および癌遺伝子が染色体に組み込まれている巨核球であって、当該外来性の遺伝子が発現していない細胞。
- 薬剤応答性で発現する外来性のp16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子がさらに染色体に組み込まれており、当該外来性遺伝子が発現していない、請求項27に記載の細胞。
- 前記アポトーシス抑制遺伝子が、BCL-XL遺伝子である、請求項27または28に記載の細胞。
- 前記癌遺伝子が、c-MYC遺伝子である、請求項27から29のいずれか1項に記載の細胞。
- 前記p16遺伝子又はp19遺伝子の発現を抑制する遺伝子、Ink4a/Arf遺伝子の発現を抑制する遺伝子及びポリコーム遺伝子から成る群より選択される1つの遺伝子が、BMI1である、請求項27から30のいずれか1項に記載の細胞。
- 巨核球の製造に適した造血前駆細胞を選択する方法であって、KLF1の発現またはFLI1の発現を測定する工程を含む方法。
- 前記KLF1の発現が低い造血前駆細胞を選択する工程を含む、請求項32に記載の方法。
- 前記FLI1の発現が高い造血前駆細胞を選択する工程を含む、請求項33に記載の方法。
- 前記造血前駆細胞が、多能性幹細胞から分化誘導された細胞である、請求項32から33のいずれか1項に記載の方法。
- 以下の工程を含む巨核球製造に適した多能性幹細胞を選択する方法;
(i)多能性幹細胞から造血幹細胞を製造する工程、および、
(ii)工程(i)で製造された造血前駆細胞において、KLF1の発現およびFLI1の発現の測定する工程。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14749207.8A EP2955223B1 (en) | 2013-02-08 | 2014-02-10 | Production methods for megakaryocytes and platelets |
| JP2014560832A JP6495658B2 (ja) | 2013-02-08 | 2014-02-10 | 巨核球及び血小板の製造方法 |
| US14/763,746 US20160002599A1 (en) | 2013-02-08 | 2014-02-10 | Production methods for megakaryocytes and platelets |
| US17/395,552 US20220017866A1 (en) | 2013-02-08 | 2021-08-06 | Production methods for megakaryocytes and platelets |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013023013 | 2013-02-08 | ||
| JP2013-023013 | 2013-02-08 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/763,746 A-371-Of-International US20160002599A1 (en) | 2013-02-08 | 2014-02-10 | Production methods for megakaryocytes and platelets |
| US17/395,552 Continuation US20220017866A1 (en) | 2013-02-08 | 2021-08-06 | Production methods for megakaryocytes and platelets |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014123242A1 true WO2014123242A1 (ja) | 2014-08-14 |
Family
ID=51299832
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/053087 WO2014123242A1 (ja) | 2013-02-08 | 2014-02-10 | 巨核球及び血小板の製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (2) | US20160002599A1 (ja) |
| EP (1) | EP2955223B1 (ja) |
| JP (1) | JP6495658B2 (ja) |
| WO (1) | WO2014123242A1 (ja) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016129593A1 (ja) * | 2015-02-10 | 2016-08-18 | 国立大学法人京都大学 | 血小板の機能を維持および/または増強するための組成物 |
| WO2016143836A1 (ja) * | 2015-03-09 | 2016-09-15 | 株式会社メガカリオン | 巨核球を含む培養物の製造方法及びこれを用いた血小板の製造方法 |
| WO2017047492A1 (ja) * | 2015-09-15 | 2017-03-23 | 株式会社メガカリオン | 回転式撹拌培養法による血小板の製造方法 |
| WO2017077964A1 (ja) * | 2015-11-02 | 2017-05-11 | 株式会社メガカリオン | 往復動撹拌装置を用いた血小板の製造方法 |
| WO2017131228A1 (ja) | 2016-01-29 | 2017-08-03 | 国立大学法人京都大学 | 血小板産生促進剤のスクリーニング方法 |
| WO2017131230A1 (ja) | 2016-01-29 | 2017-08-03 | 国立大学法人京都大学 | 血小板産生促進剤及びそれを用いた血小板の製造方法 |
| WO2018052126A1 (ja) * | 2016-09-16 | 2018-03-22 | 国立大学法人京都大学 | 巨核球細胞群における細胞の不均質性を識別する方法及び血小板の製造方法 |
| WO2018164040A1 (ja) | 2017-03-06 | 2018-09-13 | 国立大学法人京都大学 | 血小板の製造方法 |
| EP3296390A4 (en) * | 2015-04-14 | 2019-01-09 | Kyoto University | METHOD FOR THE PRODUCTION OF STEM CELL CLONES FOR INDUCING DIFFERENTIATION IN BODILY CELLS |
| WO2019009364A1 (ja) | 2017-07-07 | 2019-01-10 | 国立大学法人京都大学 | 血小板の製造方法および製造装置、ならびに血小板の製造装置における運転条件の決定方法 |
| WO2019059234A1 (ja) | 2017-09-19 | 2019-03-28 | 株式会社メガカリオン | 血小板の製造方法、血小板製剤の製造方法、および血液製剤の製造方法 |
| WO2019059235A1 (ja) | 2017-09-19 | 2019-03-28 | 株式会社メガカリオン | 精製血小板の製造方法、血小板製剤の製造方法、血液製剤の製造方法、血小板保存液、血小板保存剤および血小板の保存方法 |
| WO2019124348A1 (ja) * | 2017-12-19 | 2019-06-27 | 国立大学法人京都大学 | 新規骨分化誘導方法 |
| WO2020184685A1 (ja) * | 2019-03-13 | 2020-09-17 | 株式会社メガカリオン | 血球減少症のモデル動物の製造方法、血球減少症モデル動物、血球機能の評価方法、血球の製造方法、血球減少症の治療薬候補物質のスクリーニング方法、および血球減少症の治療薬の候補物質の製造方法 |
| WO2021117900A1 (ja) | 2019-12-13 | 2021-06-17 | 株式会社メガカリオン | 組成物およびその用途 |
| WO2021117886A1 (ja) | 2019-12-12 | 2021-06-17 | 国立大学法人千葉大学 | 巨核球および血小板を含む凍結乾燥製剤 |
| WO2022092169A1 (ja) | 2020-10-27 | 2022-05-05 | 国立大学法人 長崎大学 | 骨形成組成物およびその用途 |
| WO2022265117A1 (ja) | 2021-06-18 | 2022-12-22 | 株式会社メガカリオン | 血小板産生能が増強された多核化巨核球細胞の製造方法、血小板の製造方法、血小板製剤の製造方法、および血液製剤の製造方法 |
| WO2023277153A1 (ja) | 2021-06-30 | 2023-01-05 | 国立大学法人千葉大学 | 骨髄系共通前駆細胞(cmp)又は骨髄球系前駆細胞の増殖性を向上させる方法 |
| US11952587B2 (en) | 2017-03-06 | 2024-04-09 | Kyoto University | Method for producing platelets |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201210857D0 (en) | 2012-06-19 | 2012-08-01 | Cambridge Entpr Ltd | Transcription factor mediated programming towards megakaryocytes |
| ES2896354T3 (es) | 2012-12-21 | 2022-02-24 | Astellas Inst For Regenerative Medicine | Métodos para la producción de plaquetas a partir de células madre pluripotentes |
| CN107208058B (zh) * | 2015-01-26 | 2021-06-25 | 宇部兴产株式会社 | 利用骨髓类似结构的细胞培养方法、和用于骨损伤部位的治疗的聚酰亚胺多孔膜 |
| WO2020185856A1 (en) * | 2019-03-11 | 2020-09-17 | The Children's Medical Center Corporation | Methods for increasing platelet production |
| CN110592017B (zh) * | 2019-07-12 | 2021-02-19 | 中国医学科学院血液病医院(中国医学科学院血液学研究所) | 组蛋白甲基转移酶抑制剂在制备促进巨核细胞增殖或血小板产生的产品中的用途 |
| EP4112720A4 (en) * | 2020-02-28 | 2024-02-28 | Otsuka Pharma Co Ltd | GENETICALLY MODIFIED MEGAKARYOCYTE, MODIFIED PLATELETS AND METHOD FOR PRODUCING THE GENETICALLY MODIFIED MEGAKARYOCYTES AND THE MODIFIED PLATELETS |
Citations (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| JP2002526093A (ja) * | 1998-10-07 | 2002-08-20 | アイシス・ファーマシューティカルス・インコーポレーテッド | bcl−x発現のアンチセンスモジュレーション |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| WO2008041370A1 (fr) | 2006-10-04 | 2008-04-10 | The University Of Tokyo | Structure renfermant des cellules progénitrices hématopoïétiques issues de cellules es et procédé de préparation de cellules sanguines faisant appel à ladite structure |
| WO2008118820A2 (en) | 2007-03-23 | 2008-10-02 | Wisconsin Alumni Research Foundation | Somatic cell reprogramming |
| WO2009007852A2 (en) | 2007-06-15 | 2009-01-15 | Izumi Bio, Inc | Multipotent/pluripotent cells and methods |
| WO2009032194A1 (en) | 2007-08-31 | 2009-03-12 | Whitehead Institute For Biomedical Research | Wnt pathway stimulation in reprogramming somatic cells |
| WO2009057831A1 (ja) | 2007-10-31 | 2009-05-07 | Kyoto University | 核初期化方法 |
| WO2009058413A1 (en) | 2007-10-29 | 2009-05-07 | Shi-Lung Lin | Generation of human embryonic stem-like cells using intronic rna |
| WO2009075119A1 (ja) | 2007-12-10 | 2009-06-18 | Kyoto University | 効率的な核初期化方法 |
| WO2009079007A1 (en) | 2007-12-17 | 2009-06-25 | Gliamed, Inc. | Stem-like cells and method for reprogramming adult mammalian somatic cells |
| WO2009091659A2 (en) | 2008-01-16 | 2009-07-23 | Shi-Lung Lin | Generation of tumor-free embryonic stem-like pluripotent cells using inducible recombinant rna agents |
| WO2009101084A1 (en) | 2008-02-13 | 2009-08-20 | Fondazione Telethon | Method for reprogramming differentiated cells |
| WO2009102983A2 (en) | 2008-02-15 | 2009-08-20 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| WO2009101407A2 (en) | 2008-02-11 | 2009-08-20 | Cambridge Enterprise Limited | Improved reprogramming of mammalian cells, and the cells obtained |
| WO2009114949A1 (en) | 2008-03-20 | 2009-09-24 | UNIVERSITé LAVAL | Methods for deprogramming somatic cells and uses thereof |
| WO2009117439A2 (en) | 2008-03-17 | 2009-09-24 | The Scripps Research Institute | Combined chemical and genetic approaches for generation of induced pluripotent stem cells |
| WO2009123349A1 (ja) | 2008-03-31 | 2009-10-08 | オリエンタル酵母工業株式会社 | 多能性幹細胞を増殖させる方法 |
| WO2009122747A1 (ja) | 2008-04-01 | 2009-10-08 | 国立大学法人東京大学 | iPS細胞からの血小板の調製方法 |
| WO2009126251A2 (en) | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| WO2009157593A1 (en) | 2008-06-27 | 2009-12-30 | Kyoto University | Method of efficiently establishing induced pluripotent stem cells |
| WO2010009015A2 (en) | 2008-07-14 | 2010-01-21 | Oklahoma Medical Research Foundation | Production of pluripotent cells through inhibition of bright/arid3a function |
| WO2010008054A1 (ja) | 2008-07-16 | 2010-01-21 | ディナベック株式会社 | 染色体非組み込み型ウイルスベクターを用いてリプログラムされた細胞を製造する方法 |
| WO2010013845A1 (en) | 2008-07-30 | 2010-02-04 | Kyoto University | Method of efficiently establishing induced pluripotent stem cells |
| WO2010033906A2 (en) | 2008-09-19 | 2010-03-25 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| WO2010033920A2 (en) | 2008-09-19 | 2010-03-25 | Whitehead Institute For Biomedical Research | Compositions and methods for enhancing cell reprogramming |
| WO2010042800A1 (en) | 2008-10-10 | 2010-04-15 | Nevada Cancer Institute | Methods of reprogramming somatic cells and methods of use for such cells |
| WO2010050626A1 (en) | 2008-10-30 | 2010-05-06 | Kyoto University | Method for producing induced pluripotent stem cells |
| WO2010056831A2 (en) | 2008-11-12 | 2010-05-20 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| WO2010068955A2 (en) | 2008-12-13 | 2010-06-17 | Dna Microarray | MICROENVIRONMENT NICHE ASSAY FOR CiPS SCREENING |
| WO2010098419A1 (en) | 2009-02-27 | 2010-09-02 | Kyoto University | Novel nuclear reprogramming substance |
| WO2010102267A2 (en) | 2009-03-06 | 2010-09-10 | Ipierian, Inc. | Tgf-beta pathway inhibitors for enhancement of cellular reprogramming of human cells |
| WO2010111409A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Pluripotent stem cells |
| WO2010111422A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Induced pluripotent stem cell generation using two factors and p53 inactivation |
| WO2010115050A2 (en) | 2009-04-01 | 2010-10-07 | The Regents Of The University Of California | Embryonic stem cell specific micrornas promote induced pluripotency |
| WO2010124290A2 (en) | 2009-04-24 | 2010-10-28 | Whitehead Institute For Biomedical Research | Compositions and methods for deriving or culturing pluripotent cells |
| WO2010137746A1 (en) | 2009-05-29 | 2010-12-02 | Kyoto University | Method for producing induced pluripotent stem cells and method for culturing the same |
| WO2010147612A1 (en) | 2009-06-18 | 2010-12-23 | Lixte Biotechnology, Inc. | Methods of modulating cell regulation by inhibiting p53 |
| WO2010147395A2 (en) | 2009-06-16 | 2010-12-23 | Korea Research Institute Of Bioscience And Biotechnology | Medium composition comprising neuropeptide y for the generation, maintenance, prologned undifferentiated growth of pluripotent stem cells and method of culturing pluripotent stem cell using the same |
| WO2011007900A1 (ja) | 2009-07-15 | 2011-01-20 | Dezawa Mari | 生体組織から単離できる多能性幹細胞 |
| WO2011034073A1 (ja) | 2009-09-15 | 2011-03-24 | 国立大学法人東京大学 | 分化細胞の新規製造法 |
| WO2012157586A1 (ja) | 2011-05-13 | 2012-11-22 | 国立大学法人東京大学 | 多核化巨核球細胞、及び血小板の製造方法 |
| WO2013163296A1 (en) | 2012-04-24 | 2013-10-31 | The Brigham And Women's Hospital, Inc. | Generating pluripotent cells de novo |
-
2014
- 2014-02-10 US US14/763,746 patent/US20160002599A1/en not_active Abandoned
- 2014-02-10 WO PCT/JP2014/053087 patent/WO2014123242A1/ja active Application Filing
- 2014-02-10 JP JP2014560832A patent/JP6495658B2/ja active Active
- 2014-02-10 EP EP14749207.8A patent/EP2955223B1/en active Active
-
2021
- 2021-08-06 US US17/395,552 patent/US20220017866A1/en active Pending
Patent Citations (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| JP2002526093A (ja) * | 1998-10-07 | 2002-08-20 | アイシス・ファーマシューティカルス・インコーポレーテッド | bcl−x発現のアンチセンスモジュレーション |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| WO2008041370A1 (fr) | 2006-10-04 | 2008-04-10 | The University Of Tokyo | Structure renfermant des cellules progénitrices hématopoïétiques issues de cellules es et procédé de préparation de cellules sanguines faisant appel à ladite structure |
| WO2008118820A2 (en) | 2007-03-23 | 2008-10-02 | Wisconsin Alumni Research Foundation | Somatic cell reprogramming |
| WO2009007852A2 (en) | 2007-06-15 | 2009-01-15 | Izumi Bio, Inc | Multipotent/pluripotent cells and methods |
| WO2009032194A1 (en) | 2007-08-31 | 2009-03-12 | Whitehead Institute For Biomedical Research | Wnt pathway stimulation in reprogramming somatic cells |
| WO2009058413A1 (en) | 2007-10-29 | 2009-05-07 | Shi-Lung Lin | Generation of human embryonic stem-like cells using intronic rna |
| WO2009057831A1 (ja) | 2007-10-31 | 2009-05-07 | Kyoto University | 核初期化方法 |
| WO2009075119A1 (ja) | 2007-12-10 | 2009-06-18 | Kyoto University | 効率的な核初期化方法 |
| WO2009079007A1 (en) | 2007-12-17 | 2009-06-25 | Gliamed, Inc. | Stem-like cells and method for reprogramming adult mammalian somatic cells |
| WO2009091659A2 (en) | 2008-01-16 | 2009-07-23 | Shi-Lung Lin | Generation of tumor-free embryonic stem-like pluripotent cells using inducible recombinant rna agents |
| WO2009101407A2 (en) | 2008-02-11 | 2009-08-20 | Cambridge Enterprise Limited | Improved reprogramming of mammalian cells, and the cells obtained |
| WO2009101084A1 (en) | 2008-02-13 | 2009-08-20 | Fondazione Telethon | Method for reprogramming differentiated cells |
| WO2009102983A2 (en) | 2008-02-15 | 2009-08-20 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| WO2009117439A2 (en) | 2008-03-17 | 2009-09-24 | The Scripps Research Institute | Combined chemical and genetic approaches for generation of induced pluripotent stem cells |
| WO2009114949A1 (en) | 2008-03-20 | 2009-09-24 | UNIVERSITé LAVAL | Methods for deprogramming somatic cells and uses thereof |
| WO2009123349A1 (ja) | 2008-03-31 | 2009-10-08 | オリエンタル酵母工業株式会社 | 多能性幹細胞を増殖させる方法 |
| WO2009122747A1 (ja) | 2008-04-01 | 2009-10-08 | 国立大学法人東京大学 | iPS細胞からの血小板の調製方法 |
| WO2009126251A2 (en) | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| WO2009126655A2 (en) | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of a small molecule modulator |
| WO2009126250A2 (en) | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through rna interference |
| WO2009157593A1 (en) | 2008-06-27 | 2009-12-30 | Kyoto University | Method of efficiently establishing induced pluripotent stem cells |
| WO2010009015A2 (en) | 2008-07-14 | 2010-01-21 | Oklahoma Medical Research Foundation | Production of pluripotent cells through inhibition of bright/arid3a function |
| WO2010008054A1 (ja) | 2008-07-16 | 2010-01-21 | ディナベック株式会社 | 染色体非組み込み型ウイルスベクターを用いてリプログラムされた細胞を製造する方法 |
| WO2010013845A1 (en) | 2008-07-30 | 2010-02-04 | Kyoto University | Method of efficiently establishing induced pluripotent stem cells |
| WO2010033906A2 (en) | 2008-09-19 | 2010-03-25 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| WO2010033920A2 (en) | 2008-09-19 | 2010-03-25 | Whitehead Institute For Biomedical Research | Compositions and methods for enhancing cell reprogramming |
| WO2010042800A1 (en) | 2008-10-10 | 2010-04-15 | Nevada Cancer Institute | Methods of reprogramming somatic cells and methods of use for such cells |
| WO2010050626A1 (en) | 2008-10-30 | 2010-05-06 | Kyoto University | Method for producing induced pluripotent stem cells |
| WO2010056831A2 (en) | 2008-11-12 | 2010-05-20 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| WO2010068955A2 (en) | 2008-12-13 | 2010-06-17 | Dna Microarray | MICROENVIRONMENT NICHE ASSAY FOR CiPS SCREENING |
| WO2010098419A1 (en) | 2009-02-27 | 2010-09-02 | Kyoto University | Novel nuclear reprogramming substance |
| WO2010102267A2 (en) | 2009-03-06 | 2010-09-10 | Ipierian, Inc. | Tgf-beta pathway inhibitors for enhancement of cellular reprogramming of human cells |
| WO2010111409A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Pluripotent stem cells |
| WO2010111422A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Induced pluripotent stem cell generation using two factors and p53 inactivation |
| WO2010115050A2 (en) | 2009-04-01 | 2010-10-07 | The Regents Of The University Of California | Embryonic stem cell specific micrornas promote induced pluripotency |
| WO2010124290A2 (en) | 2009-04-24 | 2010-10-28 | Whitehead Institute For Biomedical Research | Compositions and methods for deriving or culturing pluripotent cells |
| WO2010137746A1 (en) | 2009-05-29 | 2010-12-02 | Kyoto University | Method for producing induced pluripotent stem cells and method for culturing the same |
| WO2010147395A2 (en) | 2009-06-16 | 2010-12-23 | Korea Research Institute Of Bioscience And Biotechnology | Medium composition comprising neuropeptide y for the generation, maintenance, prologned undifferentiated growth of pluripotent stem cells and method of culturing pluripotent stem cell using the same |
| WO2010147612A1 (en) | 2009-06-18 | 2010-12-23 | Lixte Biotechnology, Inc. | Methods of modulating cell regulation by inhibiting p53 |
| WO2011007900A1 (ja) | 2009-07-15 | 2011-01-20 | Dezawa Mari | 生体組織から単離できる多能性幹細胞 |
| WO2011034073A1 (ja) | 2009-09-15 | 2011-03-24 | 国立大学法人東京大学 | 分化細胞の新規製造法 |
| WO2012157586A1 (ja) | 2011-05-13 | 2012-11-22 | 国立大学法人東京大学 | 多核化巨核球細胞、及び血小板の製造方法 |
| WO2013163296A1 (en) | 2012-04-24 | 2013-10-31 | The Brigham And Women's Hospital, Inc. | Generating pluripotent cells de novo |
Non-Patent Citations (85)
| Title |
|---|
| BRUMMELKAMP TR ET AL., SCIENCE, vol. 296, 2002, pages 550 - 553 |
| CELL, vol. 126, 2006, pages 663 - 676 |
| CELL, vol. 131, 2007, pages 861 - 872 |
| CHADWICK ET AL., BLOOD, vol. 102, 2003, pages 906 - 15 |
| DE CUYPER, IM ET AL., BLOOD, vol. 121, 2013, pages E70 - 80 |
| DECKWERTH ET AL., DRUG DEV. RES., vol. 52, 2001, pages 579 - 586 |
| DORE LOUIS C. ET AL.: "Transcription factor networks in erythroid cell and megakaryocyte development", BLOOD, vol. 118, no. 2, 14 July 2011 (2011-07-14), pages 231 - 239, XP055272494, DOI: 10.1182/BLOOD-2011-04-285981 * |
| E. KROON ET AL., NAT. BIOTECHNOL., vol. 26, 2008, pages 443 - 452 |
| EMINLI S ET AL., STEM CELLS, vol. 26, 2008, pages 2467 - 2474 |
| FENG B ET AL., NAT CELL BIOL., vol. 11, 2009, pages 197 - 203 |
| FUHRKEN ET AL., J. BIOL. CHEM., vol. 283, 2008, pages 15589 - 15600 |
| GULLIKSSON, H. ET AL., TRANSFUSION, vol. 32, 1992, pages 435 - 440 |
| H. KAWASAKI ET AL., PROC. NATL. ACAD. SCI. USA, vol. 99, 2002, pages 1580 - 1585 |
| H. SUEMORI ET AL., BIOCHEM. BIOPHYS. RES. COMMUN., vol. 345, 2006, pages 926 - 932 |
| H. SUEMORI ET AL., DEV. DYN., vol. 222, 2001, pages 273 - 279 |
| HAN ET AL., J. BIOL. CHEM., vol. 277, 2002, pages 30128 - 30136 |
| HAN J ET AL., NATURE, vol. 463, 2010, pages 1096 - 100 |
| HENG JC ET AL., CELL STEM CELL, vol. 6, 2010, pages 167 - 74 |
| HOGLEN ET AL., J. PHARMACOL. EXP. THER, vol. 297, 2001, pages 811 - 818 |
| HOGLEN ET AL., J. PHARMACOL. EXP. THER, vol. 309, 2004, pages 634 - 640 |
| HOTCHKISS ET AL., NAT. IMMUNOL, vol. 1, 2000, pages 496 - 501 |
| HUANGFU D ET AL., NAT BIOTECHNOL., vol. 26, 2008, pages 1269 - 1275 |
| HUANGFU D ET AL., NAT. BIOTECHNOL., vol. 26, 2008, pages 795 - 797 |
| ICHIDA JK ET AL., CELL STEM CELL., vol. 5, 2009, pages 491 - 503 |
| ISABEL ET AL., BIOORG. MED. CHEM. LETT, vol. 13, 2003, pages 2137 - 2140 |
| J. A. THOMSON ET AL., BIOL. REPROD., vol. 55, 1996, pages 254 - 259 |
| J. A. THOMSON ET AL., SCIENCE, vol. 282, 1998, pages 1145 - 1147 |
| J. B. CIBELLI ET AL., NATURE BIOTECHNOL., vol. 16, 1998, pages 642 - 646 |
| J. BYRNE ET AL., NATURE, vol. 450, 2007, pages 497 - 502 |
| J. YU ET AL., SCIENCE, vol. 318, 2007, pages 1917 - 1920 |
| J.A. THOMSON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 92, 1995, pages 7844 - 7848 |
| J.A. THOMSON; V. S. MARSHALL, CURR. TOP. DEV. BIOL., vol. 38, 1998, pages 133 - 165 |
| J.L. RESNICK ET AL., NATURE, vol. 359, 1992, pages 550 - 551 |
| K. SHINOHARA ET AL., CELL, vol. 119, 2004, pages 1001 - 1012 |
| K. TAKAHASHI ET AL., CELL, vol. 131, 2007, pages 861 - 872 |
| K. TAKAHASHI; S. YAMANAKA, CELL, vol. 126, 2006, pages 663 - 676 |
| KATAGIRI T ET AL., BIOCHEM BIOPHYS RES COMMUN, vol. 172, 1990, pages 295 - 299 |
| KIM JB ET AL., NATURE, vol. 461, 2009, pages 649 - 643 |
| KIYOKA WAKAYAMA ET AL., EXPERIMENTAL MEDICINE, vol. 26, no. 5, 2008, pages 47 - 52 |
| KLIMANSKAYA I ET AL., NATURE, vol. 444, 2006, pages 481 - 485 |
| KLUTER H. ET AL.: "Impact of buffy coat storage on the generation of inflammatory cytokines and platelet activation", TRANSFUSION, vol. 37, 1 April 1997 (1997-04-01), pages 362 - 367, XP055272486, DOI: 10.1046/J.1537-2995.1997.37497265335.X * |
| KOBAYASHI, T. ET AL., CELL, vol. 142, 2010, pages 787 - 799 |
| LEUNG-TOUNG ET AL., CURR. MED. CHEM., vol. 9, 2002, pages 979 - 1002 |
| LORDIER ET AL., BLOOD, vol. 112, 2008, pages 3164 - 3174 |
| LOS ET AL., DRUG DISCOV. TODAY, vol. 8, 2003, pages 67 - 77 |
| LYSSIOTIS CA ET AL., PROC NATL ACAD SCI U S A., vol. 106, 2009, pages 8912 - 8917 |
| M. J. EVANS; M. H. KAUFMAN, NATURE, vol. 292, 1981, pages 154 - 156 |
| M. KANATSU-SHINOHARA ET AL., BIOL. REPROD., vol. 69, 2003, pages 612 - 616 |
| M. UENO ET AL., PROC. NATL. ACAD. SCI. USA, vol. 103, 2006, pages 9554 - 9559 |
| MAEKAWA M ET AL., NATURE, vol. 474, 2011, pages 225 - 9 |
| MALI P ET AL., STEM CELLS, vol. 28, 2010, pages 713 - 720 |
| MARSON A, CELL STEM CELL, vol. 3, 2008, pages 132 - 135 |
| MASANORI TAKEBAYASHI ET AL.: "Experimental Medicine", vol. 26, 2008, YODOSHA, pages: 41 - 46 |
| METHOT ET AL., J. EXP. MED., vol. 199, 2004, pages 199 - 207 |
| NAKAGAWA, M. ET AL., NAT. BIOTECHNOL., vol. 26, 2008, pages 101 - 106 |
| NIWA A ET AL., J CELL PHYSIOL., vol. 221, no. 2, November 2009 (2009-11-01), pages 367 - 77 |
| OHMINE K ET AL., ONCOGENE, vol. 20, 2001, pages 8249 - 8257 |
| OKITA K ET AL., STEM CELLS, vol. 31, 2012, pages 458 - 66 |
| PROULX ET AL., BIOTECHNOL. BIOENG., vol. 88, 2004, pages 675 - 680 |
| R.L. JUDSON ET AL., NAT. BIOTECH., vol. 27, 2009, pages 459 - 461 |
| RANDLE ET AL., EXPERT OPIN. INVESTIG. DRUGS, vol. 10, 2001, pages 1207 - 1209 |
| S. WAKAYAMA ET AL., BIOL. REPROD., vol. 72, 2005, pages 932 - 936 |
| SAEKI ET AL., STEM CELLS, vol. 27, 2009, pages 59 - 67 |
| SCHWEINFURTH ET AL., PLATELETS, vol. 21, 2010, pages 648 - 657 |
| SCIENCE, vol. 318, 2007, pages 1917 - 1920 |
| SCIENCE, vol. 322, 2008, pages 945 - 949 |
| SCIENCE, vol. 322, 2008, pages 949 - 953 |
| SCOTT ET AL., J. PHARMACOL. EXP. THER, vol. 304, 2003, pages 433 - 440 |
| SHI Y ET AL., CELL STEM CELL, vol. 2, 2008, pages 525 - 528 |
| SHI Y ET AL., CELL STEM CELL, vol. 3, 2008, pages 568 - 574 |
| SUN N ET AL., PROC NATL ACAD SCI U S A., vol. 106, 2009, pages 15720 - 15725 |
| T. WAKAYAMA ET AL., SCIENCE, vol. 292, 2001, pages 740 - 743 |
| TAKAHASHI K ET AL., CELL, vol. 131, 2007, pages 861 - 872 |
| TAKAHASHI K ET AL., PLOS ONE., vol. 4, 2009, pages E8067 |
| TAKAYAMA ET AL., BLOOD, vol. 111, 2008, pages 5298 - 5306 |
| TAKAYAMA N. ET AL., J EXP MED., 2010, pages 2817 - 2830 |
| TAKAYAMA NAOYA: "Platelet Production System Using an Immortalized Megakaryocyte Cell Line Derived From Human Pluripotent Stem Cells", AMERICAN SOCIETY OF HEMATOLOGY, 10 December 2011 (2011-12-10), XP008180270, Retrieved from the Internet <URL:http://www.reuters.com/article/2011/12/10/idUS70817+10-Dec-2011+PRN20111210> [retrieved on 20140424] * |
| THOMSON JA ET AL., PROC NATL. ACAD. SCI. U S A., vol. 92, 1995, pages 7844 - 7848 |
| THOMSON JA ET AL., SCIENCE, vol. 282, 1998, pages 1145 - 1147 |
| VIJAYARAGAVAN ET AL., CELL STEM CELL, vol. 4, 2009, pages 248 - 62 |
| VOORHOEVE PM; AGAMI R, CELL, vol. 4, 2003, pages 311 - 319 |
| WARREN L, CELL STEM CELL, vol. 7, 2010, pages 618 - 630 |
| Y. MATSUI ET AL., CELL, vol. 70, 1992, pages 841 - 847 |
| YOSHIDA Y ET AL., CELL STEM CELL, vol. 5, 2009, pages 237 - 241 |
| ZHAO Y ET AL., CELL STEM CELL, vol. 3, 2008, pages 475 - 479 |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016129593A1 (ja) * | 2015-02-10 | 2016-08-18 | 国立大学法人京都大学 | 血小板の機能を維持および/または増強するための組成物 |
| JPWO2016143836A1 (ja) * | 2015-03-09 | 2017-12-21 | 株式会社メガカリオン | 巨核球を含む培養物の製造方法及びこれを用いた血小板の製造方法 |
| WO2016143836A1 (ja) * | 2015-03-09 | 2016-09-15 | 株式会社メガカリオン | 巨核球を含む培養物の製造方法及びこれを用いた血小板の製造方法 |
| EP3296390A4 (en) * | 2015-04-14 | 2019-01-09 | Kyoto University | METHOD FOR THE PRODUCTION OF STEM CELL CLONES FOR INDUCING DIFFERENTIATION IN BODILY CELLS |
| RU2730034C2 (ru) * | 2015-04-14 | 2020-08-14 | Киото Юниверсити | Способ получения клона стволовых клеток, пригодного для индуцирования дифференцировки в соматические клетки |
| CN108026511A (zh) * | 2015-09-15 | 2018-05-11 | 株式会社美加细胞 | 利用旋转式搅拌培养法的血小板的制造方法 |
| US10570372B2 (en) | 2015-09-15 | 2020-02-25 | Megakaryon Corporation | Method for manufacturing platelets by rotary stirring culture method |
| KR102641631B1 (ko) * | 2015-09-15 | 2024-02-29 | 가부시키가이샤 메가카리온 | 회전식 교반 배양법에 의한 혈소판의 제조 방법 |
| KR20180044427A (ko) * | 2015-09-15 | 2018-05-02 | 가부시키가이샤 메가카리온 | 회전식 교반 배양법에 의한 혈소판의 제조 방법 |
| JPWO2017047492A1 (ja) * | 2015-09-15 | 2018-07-05 | 株式会社メガカリオン | 回転式撹拌培養法による血小板の製造方法 |
| WO2017047492A1 (ja) * | 2015-09-15 | 2017-03-23 | 株式会社メガカリオン | 回転式撹拌培養法による血小板の製造方法 |
| WO2017077964A1 (ja) * | 2015-11-02 | 2017-05-11 | 株式会社メガカリオン | 往復動撹拌装置を用いた血小板の製造方法 |
| CN108473954A (zh) * | 2015-11-02 | 2018-08-31 | 株式会社美加细胞 | 使用往复运动搅拌装置的血小板的制造方法 |
| JPWO2017077964A1 (ja) * | 2015-11-02 | 2018-10-11 | 株式会社メガカリオン | 往復動撹拌装置を用いた血小板の製造方法 |
| WO2017131230A1 (ja) | 2016-01-29 | 2017-08-03 | 国立大学法人京都大学 | 血小板産生促進剤及びそれを用いた血小板の製造方法 |
| WO2017131228A1 (ja) | 2016-01-29 | 2017-08-03 | 国立大学法人京都大学 | 血小板産生促進剤のスクリーニング方法 |
| WO2018052126A1 (ja) * | 2016-09-16 | 2018-03-22 | 国立大学法人京都大学 | 巨核球細胞群における細胞の不均質性を識別する方法及び血小板の製造方法 |
| WO2018164040A1 (ja) | 2017-03-06 | 2018-09-13 | 国立大学法人京都大学 | 血小板の製造方法 |
| US11952587B2 (en) | 2017-03-06 | 2024-04-09 | Kyoto University | Method for producing platelets |
| JPWO2018164040A1 (ja) * | 2017-03-06 | 2019-12-26 | 国立大学法人京都大学 | 血小板の製造方法 |
| JP7297203B2 (ja) | 2017-03-06 | 2023-06-26 | 国立大学法人京都大学 | 血小板の製造方法 |
| WO2019009364A1 (ja) | 2017-07-07 | 2019-01-10 | 国立大学法人京都大学 | 血小板の製造方法および製造装置、ならびに血小板の製造装置における運転条件の決定方法 |
| WO2019059234A1 (ja) | 2017-09-19 | 2019-03-28 | 株式会社メガカリオン | 血小板の製造方法、血小板製剤の製造方法、および血液製剤の製造方法 |
| US11773374B2 (en) | 2017-09-19 | 2023-10-03 | Megakaryon Corporation | Method for producing purified platelets, method for producing platelet product, method for producing blood product, platelet preserving solution, platelet preserving agent, and method for preserving platelets |
| WO2019059235A1 (ja) | 2017-09-19 | 2019-03-28 | 株式会社メガカリオン | 精製血小板の製造方法、血小板製剤の製造方法、血液製剤の製造方法、血小板保存液、血小板保存剤および血小板の保存方法 |
| JPWO2019124348A1 (ja) * | 2017-12-19 | 2020-12-03 | 国立大学法人京都大学 | 新規骨分化誘導方法 |
| US11859209B2 (en) | 2017-12-19 | 2024-01-02 | Kyoto University | Method for inducing osteogenic differentiation |
| WO2019124348A1 (ja) * | 2017-12-19 | 2019-06-27 | 国立大学法人京都大学 | 新規骨分化誘導方法 |
| WO2020184685A1 (ja) * | 2019-03-13 | 2020-09-17 | 株式会社メガカリオン | 血球減少症のモデル動物の製造方法、血球減少症モデル動物、血球機能の評価方法、血球の製造方法、血球減少症の治療薬候補物質のスクリーニング方法、および血球減少症の治療薬の候補物質の製造方法 |
| WO2021117886A1 (ja) | 2019-12-12 | 2021-06-17 | 国立大学法人千葉大学 | 巨核球および血小板を含む凍結乾燥製剤 |
| WO2021117900A1 (ja) | 2019-12-13 | 2021-06-17 | 株式会社メガカリオン | 組成物およびその用途 |
| WO2022092169A1 (ja) | 2020-10-27 | 2022-05-05 | 国立大学法人 長崎大学 | 骨形成組成物およびその用途 |
| WO2022265117A1 (ja) | 2021-06-18 | 2022-12-22 | 株式会社メガカリオン | 血小板産生能が増強された多核化巨核球細胞の製造方法、血小板の製造方法、血小板製剤の製造方法、および血液製剤の製造方法 |
| WO2023277153A1 (ja) | 2021-06-30 | 2023-01-05 | 国立大学法人千葉大学 | 骨髄系共通前駆細胞(cmp)又は骨髄球系前駆細胞の増殖性を向上させる方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2955223A4 (en) | 2016-07-27 |
| JP6495658B2 (ja) | 2019-04-03 |
| JPWO2014123242A1 (ja) | 2017-02-02 |
| US20160002599A1 (en) | 2016-01-07 |
| US20220017866A1 (en) | 2022-01-20 |
| EP2955223A1 (en) | 2015-12-16 |
| EP2955223B1 (en) | 2019-12-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6495658B2 (ja) | 巨核球及び血小板の製造方法 | |
| EP2794860B1 (en) | Method for inducing differentiation of human pluripotent stem cells into intermediate mesoderm cells | |
| US8652845B2 (en) | Method for producing mesodermal cells by culturing under adherent conditions and without co-culture with cells from a different species in a serum-free medium | |
| JP6738572B2 (ja) | 肺胞上皮細胞の分化誘導法 | |
| US9822342B2 (en) | Method of efficiently inducing cardiomyocytes | |
| JPWO2014168264A1 (ja) | 肺胞上皮前駆細胞の誘導方法 | |
| US20170130202A1 (en) | Methods respectively for producing mesodermal cells and hematopoietic cells | |
| JP2012532585A (ja) | 多能性幹細胞から骨格筋前駆細胞への分化誘導方法 | |
| JP7357369B2 (ja) | 新規腎前駆細胞マーカーおよびそれを利用した腎前駆細胞の濃縮方法 | |
| JP5995247B2 (ja) | 多能性幹細胞から樹状細胞を製造する方法 | |
| JP7274683B2 (ja) | 多能性幹細胞から樹状分岐した集合管を伴う腎臓構造を作製する方法 | |
| WO2021085462A1 (ja) | 多能性幹細胞から造血性内皮細胞および/または造血前駆細胞を製造する方法 | |
| WO2016148307A1 (ja) | 気道上皮細胞の分化誘導法 | |
| JP6385340B2 (ja) | 巨核球の成熟化促進物質 | |
| JP7078934B2 (ja) | 特定のラミニン上での多能性幹細胞の培養方法 | |
| JP6873898B2 (ja) | 体細胞への分化誘導に適した幹細胞クローンを製造する方法 | |
| WO2022014604A1 (ja) | 骨格筋前駆細胞及びその精製方法、筋原性疾患を治療するための組成物、並びに骨格筋前駆細胞を含む細胞群の製造方法 | |
| WO2017073794A1 (ja) | 多能性幹細胞から3次元の心筋組織を製造する方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14749207 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014560832 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014749207 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14763746 Country of ref document: US |